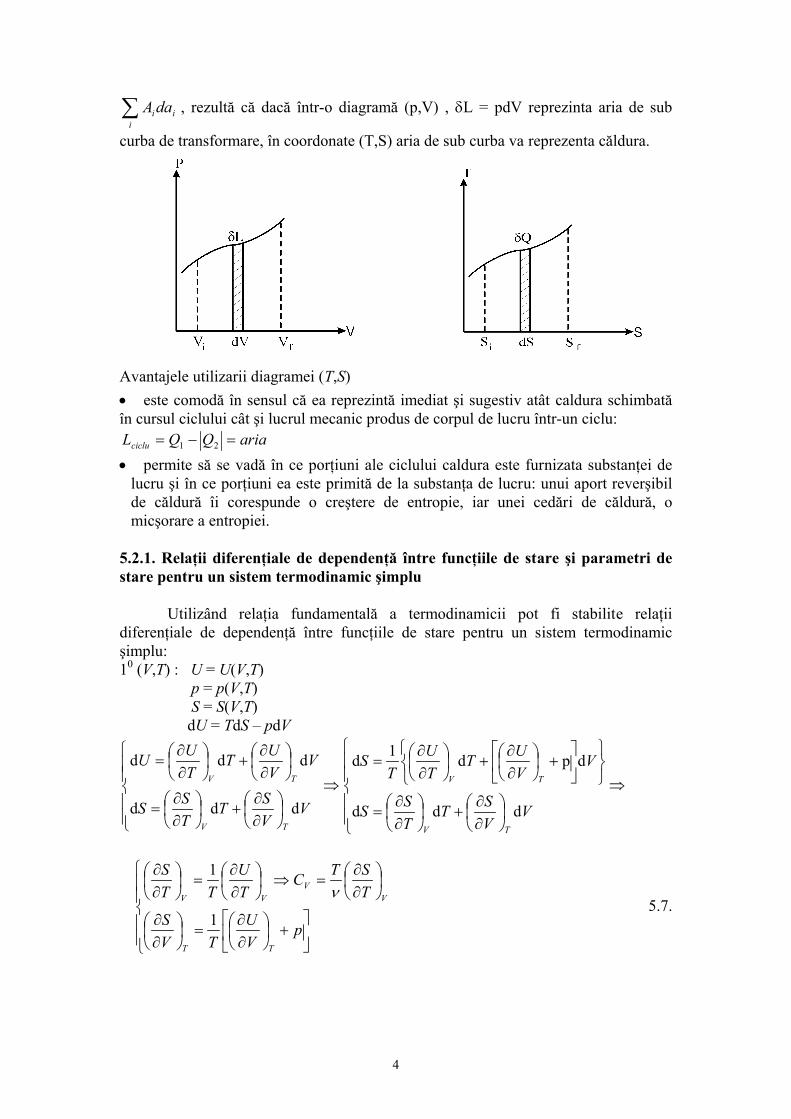
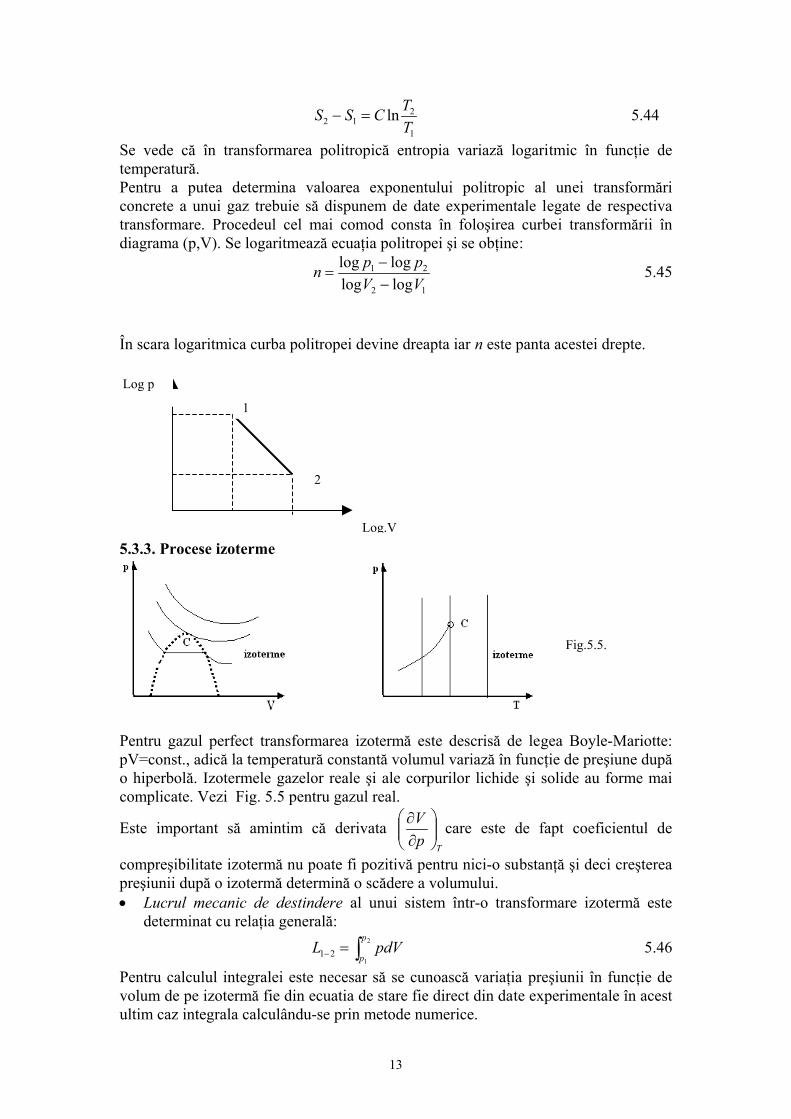
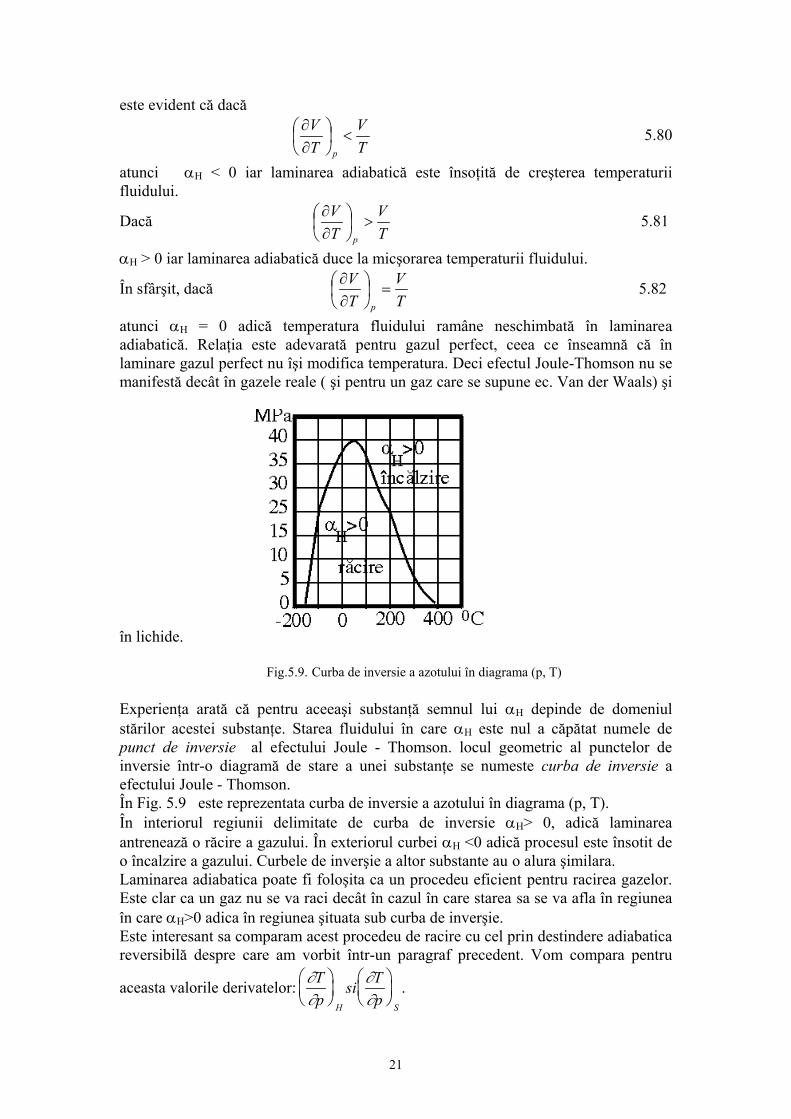
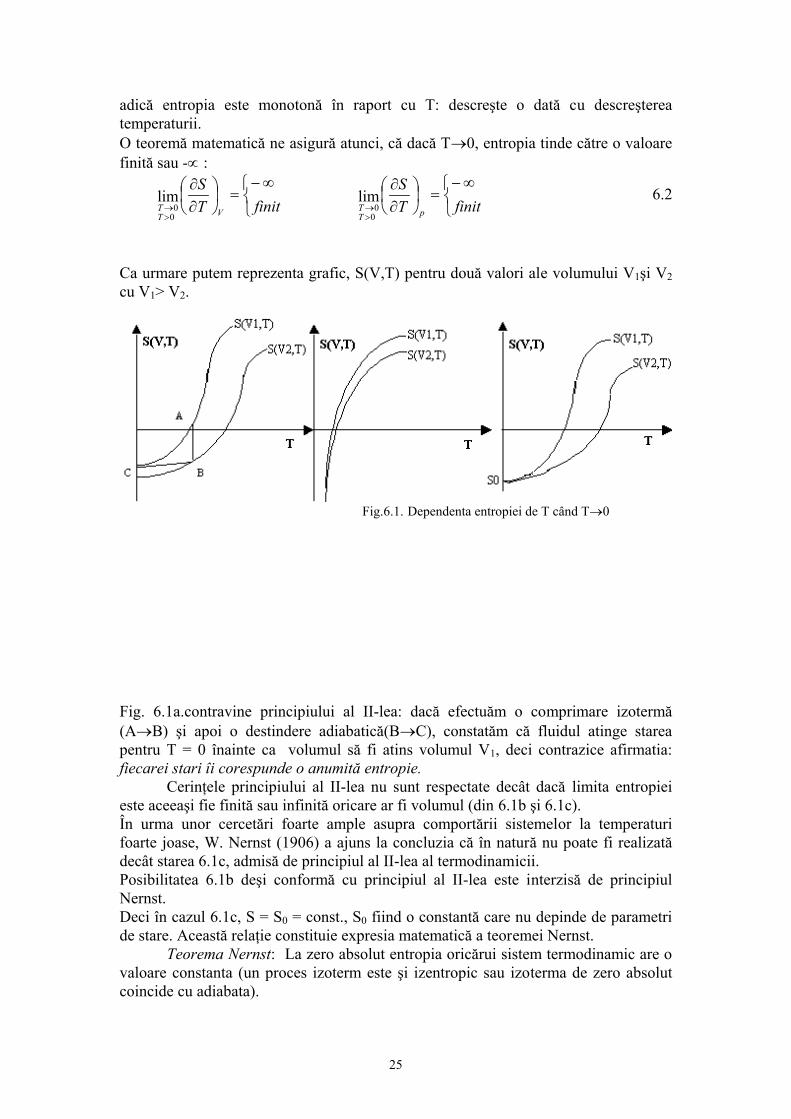
Prof.univ.dr.Sabina Ştefan Principiul ştiinţei, aproape definiţia ei este: proba oricarei cunoaşteri este experienţa Experienţa este singurul judecător al “adevărului” ştiinţific. 1. INTRODUCERE 1.1.Fizica moleculară: obiect şi metode de studiu. Studiind la mecanică legile care guvernează mişcarea corpurilor, nu ne-am pus problema cum sunt structurate corpurile şi care sunt proprietaţile lor intrinseci. Masa şi dimensiunile corpurilor erau suficiente pentru studiul mişcarii, aplicarea forţelor asupra corpului necesitând cunoaşterea numai a acestor proprietaţi. Este evident însa că numai prin masă sau dimensiuni nu pot fi caracterizate în întregime corpurile. Proprietaţile care nu intervin în mişcarea mecanică sunt de fapt intim legate prin alte fenomene naturale. Proprietăţile corpurilor sunt funcţii de structura lor, de elementele care le compun şi de forţele de interacţie dintre aceste elemente. Problema structurii materiei este una din problemele fundamentale ale fizicii.În reprezentarile cu care noi operăm zilnic, corpurile sunt în general considerate ca un continuum, adică acestea ocupă spaţiul cu materia care le compune. Din acest punct de vedere unele procese, fenomene se explică simplu; de exemplu se ştie ca încălzirea sau răcirea unui corp determină dilatarea, respectiv contractarea acestuia. Reprezentarea corpului ca un continuum explică astfel de procese prin modificarea volumului pe care-l ocupă materia din care este constituit corpul. Gândirea trebuie însă să meargă mai departe pentru ca aceste fenomene aparent simple să le explicăm prin ceea ce se întâmplă în structura corpurilor, pentru că de fapt corpurile se compun dintr-un număr foarte mare de particule pe care nu le putem vedea la un microscop obişnuit. Aceste particule infime ale materiei sunt moleculele iar între molecule se exercită forţele de interacţiune moleculară. Această reprezentare este reprezentarea discontinuă a materiei, acceptată din antichitate, susţinută în prezent de o teorie riguroasă şi verificată în timp de mii de experienţe. Existenţa celor trei stări de agregarea a materiei: solidă lichidă şi gazoasă este o manifestare a forţelor intermoleculare. În stare lichidă şi solidă, moleculele se atrag suficient pentru a permite corpurilor să-şi conserve volumul şi respectiv forma şi volumul în cazul solidelor. În stare gazoasă, datorită forţelor de interacţiune slabe, gazul ocupă întreg volumul, oricât de mare ar fi, adică gazul este expansibil. Această proprietate de expansibilitate a gazului pune în evidenţă că moleculele sunt într-o permanentă mişcare. O serie de alte proprietăţi ale gazului arată că această mişcare continuă, se desfaşoară în mod dezordonat, haotic (aceasta înseamnă că nu există o direcţie privelegiată, preferenţială de mişcare). Această mişcare haotică a moleculelor poartă numele de “agitaţie termică”. Proprietatea moleculelor de a se găsi în stare de agitaţie termică este proprie nu numai 1
Transcript
Prof.univ.dr.Sabina Ştefan
Principiul ştiinţei, aproape definiţia ei este: proba oricarei cunoaşteri este experienţa
Experienţa este singurul judecător al “adevărului” ştiinţific.
1. INTRODUCERE 1.1.Fizica moleculară: obiect şi metode de studiu. Studiind la mecanică legile care guvernează mişcarea corpurilor, nu ne-am pus problema cum sunt structurate corpurile şi care sunt proprietaţile lor intrinseci. Masa şi dimensiunile corpurilor erau suficiente pentru studiul mişcarii, aplicarea forţelor asupra corpului necesitând cunoaşterea numai a acestor proprietaţi. Este evident însa că numai prin masă sau dimensiuni nu pot fi caracterizate în întregime corpurile. Proprietaţile care nu intervin în mişcarea mecanică sunt de fapt intim legate prin alte fenomene naturale. Proprietăţile corpurilor sunt funcţii de structura lor, de elementele care le compun şi de forţele de interacţie dintre aceste elemente. Problema structurii materiei este una din problemele fundamentale ale fizicii.În reprezentarile cu care noi operăm zilnic, corpurile sunt în general considerate ca un continuum, adică acestea ocupă spaţiul cu materia care le compune. Din acest punct de vedere unele procese, fenomene se explică simplu; de exemplu se ştie ca încălzirea sau răcirea unui corp determină dilatarea, respectiv contractarea acestuia. Reprezentarea corpului ca un continuum explică astfel de procese prin modificarea volumului pe care-l ocupă materia din care este constituit corpul. Gândirea trebuie însă să meargă mai departe pentru ca aceste fenomene aparent simple să le explicăm prin ceea ce se întâmplă în structura corpurilor, pentru că de fapt corpurile se compun dintr-un număr foarte mare de particule pe care nu le putem vedea la un microscop obişnuit. Aceste particule infime ale materiei sunt moleculele iar între molecule se exercită forţele de interacţiune moleculară. Această reprezentare este reprezentarea discontinuă a materiei, acceptată din antichitate, susţinută în prezent de o teorie riguroasă şi verificată în timp de mii de experienţe. Existenţa celor trei stări de agregarea a materiei: solidă lichidă şi gazoasă este o manifestare a forţelor intermoleculare. În stare lichidă şi solidă, moleculele se atrag suficient pentru a permite corpurilor să-şi conserve volumul şi respectiv forma şi volumul în cazul solidelor. În stare gazoasă, datorită forţelor de interacţiune slabe, gazul ocupă întreg volumul, oricât de mare ar fi, adică gazul este expansibil. Această proprietate de expansibilitate a gazului pune în evidenţă că moleculele sunt într-o permanentă mişcare. O serie de alte proprietăţi ale gazului arată că această mişcare continuă, se desfaşoară în mod dezordonat, haotic (aceasta înseamnă că nu există o direcţie privelegiată, preferenţială de mişcare). Această mişcare haotică a moleculelor poartă numele de “agitaţie termică”. Proprietatea moleculelor de a se găsi în stare de agitaţie termică este proprie nu numai
1
gazelor ci şi stărilor lichidă şi solidă cu precizarea că natura mişcării termice la acestea din urmă este diferită de cea a gazului. Aşadar, materia este compusa din particule infime (molecule) care interacţionează între ele şi se gasesc într-o stare de perpetuă mişcare dezordonată- agitaţia termică. Obiectul fizicii moleculare este sudiul proprietăţilor materiei, pornind de la faptul că aceasta este constituită dintr-un ansamblu format dintr-un număr foarte mare de particule în mişcare.Studiul sistemelor de acest fel prezintă dificultăţi mari, mai ales pentru că trebuie să se ţină seama de forţele de interacţiune dintre molecule. Totodata, o serie de proprietăţi ale substanţei, numeroase fenomene care se derulează în interiorul substanţei pot fi studiată fără o cunoaştere în detaliu a mecanismului mişcărilor moleculare, adică ne raportăm numai la mărimile macroscopice, mărimile care caracterizează substanţa în ansamblul său.În acest caz, raportarea la particule izolate este lipsita de semnificaţie fizica şi incorecta. De exemplu nu vorbim niciodată despre presiunea sau temperatura unei molecule! Aceşti parametrii se definesc prin proprietăţi macroscopice ale ansamblului de molecule. Deci, când se studiază proprietăţile materiei legate de agitaţia termică a moleculelor, se folosesc legi generale care sunt întotdeauna valabile, independent de natura mişcărilor moleculare, de interacţia şi de structura substanţei. Pentru toate fenomenele care le vom studia, fenomene de natură termică (fizica moleculară se mai numeşte uneori şi fizica fenomenelor termice), nu va fi nevoie să ţinem seama de structura atomică şi de natura cuantică a proceselor interatomice. Ca urmare, rezumând, studiul fenomenelor legate de mişcarea termică se poate face pe cale microscopică şi macroscopică. Schematizarea metodică pentru abordarea mişcării termice este:
neechilibru ireversibile Metoda fenomenologică la scară macroscopică (termodinamica) reprezintă ştiinţa experimentală bazată pe un număr mic de principii care sunt generalizări ale
2
experienţei. Ea nu face ipoteze asupra starii microscopice (structura materiei). De la principiile termodinamicii se pot obţine relaţii generale între coeficienţii calorici, călduri latente, coeficienţi electrici şi magnetici. Teoria cinetico-moleculara a materiei aplică legile mecanicii, moleculelor individuale ale sistemului, permiţând calculul valorilor numerice pentru căldurile specifice ale gazului şi înţelegerea proprietăţilor gazelor în termenii forţelor intramoleculare. Fizica statistică porneşte de la structura atomică a substanţei dar ignoră consideraţiile de detaliu ale moleculelor ca entitaţi singulare şi aplică consideraţii statistice ca să determine proprietăţile ansamblului macroscopic, constituit dintr-un număr enorm de molecule. Fizica statistică ca şi termodinamica au ca obiect studiul fenomenelor termice împreună cu fenomenele mecanice, electromagnetice şi chimice pe care le însoţesc şi ele diferă nu prin obiect, ci prin punctul de vedere din care studiază fenomenele. Aşadar, mişcarea termică (fenomenele termice) poate fi abordata în trei moduri, deja numite clasice: termodinamica, teoria cinetico-moleculară şi fizica statistică, alături de mai recenta abordare informaţională iniţiată de un inginer (Tribus) şi mai mult pentru ingineri. În lumea ştiinţei secolului XX s-a acceptat ideea că termodinamica statistică (Gibbs) şi într-o oarecare masură chiar teoria cinetico-moleculară a gazelor (Maxwell-Boltzmann) constituie părţi componente ale termodinamicii fenomenologice clasice (Carnot - Joule - Thomson - Gibbs - Helmholtz - Nernst)şi cu elemente din termodinamica fenomenologică ireversibilă (Onsager - Prigogine). Se spune uneori că dezvoltarea termodinamicii fenomenologice a precedat dezvoltarea teoriei cinetico-moleculare şi a termodinamicii statistice. Această afirmaţie nu este perfect adevărată pentru că în mare masură ele s-au dezvoltat simultan, uneori chiar în gândirea aceloraşi oameni de ştiinţa. Astfel, Helmoltz şi Clausius au folosit atât abordarea fenomenologică cât şi pe cea cinetico-moleculară. La fel Gibbs şi ar mai fi destule exemple..... Se poate spune că după 1850 termodinamica fenomenologică, teoria cinetico-moleculară şi termodinamica statistică s-au dezvoltat împreună şi au condus pâna la urmă la teoria cuantică (Max Planck). 1.2. Noţiuni fundamentale de termodinamică Termodinamica studiază proprietăţile generale ale materiei şi legile de desfăşurare ale proceselor naturale, ţinând seama de toate formele de mişcare: mecanică, electrică, magnetică, chimică şi în mod esenţial de mişcarea termică. Deoarece orice corp macroscopic aflat la o temperatură diferită de 0K “conţine” o anumită mişcare termică (de agitaţie moleculară: translaţie, vibraţie, rotaţie) rezultă că orice fenomen (mecanic, electric, chimic, biologic) va fi însoţit de o mişcare termică. De aici se vede importanţa si generalitatea ştiinţei termodinamice. Studiul fenomenelor termodinamice se face la scară macroscopică, comportarea sistemelor la echilibru fiind problema centrală a termodinamicii. Termodinamica, la fel ca mecanica şi-a dedus legile şi principiile din experienţă, în cazul sintezei inductive. Odată stabilite principiile, ea s-a dezvoltat axiomatic, deductiv (şase postulate).
3
Adecvarea la realitate a rezultatelor ei este asigurată de faptul ca principiile au fost “extrase” din experienţă (practică). Conceptele cu care operăm în termodinamică s-au preluat de la mecanică, uneori conştient alteori subconştient prin limbaj, analogii etc.. Ceea ce vreau sa subliniez este că nu este important să dăm definiţii ci este important să înţelegem despre ce vorbim, sa învăaţăm un limbaj în spatele căruia să avem semnificaţiile foarte clare. Astfel, va trebui sa fie înţelese foarte bine noţiunile de bază: sistem, stare, structura, interacţie, proces. 1.2.1. Sistem termodinamic. În tratarea oricarei probleme de termodinamică este absolut obligatoriu să se precizeze care este sistemul care face obiectul studiului. Din sistem pot face parte corpuri care pot fi pure din punct de vedere chimic sau amestecuri, dar şi câmpuri ca de exemplu cel de radiaţii, deci orice porţiune finită din Univers pentru care se poate defini un interior si un exterior. Se poate defini ca urmare, sistemul termodinamic ansamblul de entităţi macroscopice, corpuri sau câmpuri care pot schimba energie şi substanţă între ele sau cu mediul înconjurator. Sistemul termodinamic este de fapt un sistem fizic care trebuie să îndeplinească condiţiile: a) conţine un număr foarte mare dar finit de microsisteme; b) este limitat spaţial . Termodinamica studiază în fapt condiţiile şi relaţiile cantitative cele mai generale, privind schimbul de energie şi substanţă în sistemele termodinamice. Studiul sistemelor termodinamice presupune stabilirea cât mai exactă a stării lor interne cât şi a determinării lor externe în raport cu alte sisteme cu care se află în interacţie. 1.2.2. Interacţia sistem - mediul înconjurător Faptul că separăm mintal obiectele care aparţin sistemului de cele care nu îi aparţin nu înseamnă că ne închipuim sistemul ca fiind izolat de restul lumii. Dimpotrivă, în prim plan în studiul termodinamic stă interacţiunea dintre sistem şi lumea înconjurătoare. În plus, trebuie precizat că şi între parţile unui sistem există interacţii. Principiul filozofic al interacţiunii universale fiind o consecinţa a generalizarii rezultatelor ştiinţelor naturii, afirma ca nu exista nici-o “particica materială” care să nu se afle într-o acţiune reciprocă cu alte parţi materiale ale universului. Această acţiune reciprocă a tuturor sistemelor materiale este cauza tuturor modificărilor de stare şi de structură care au loc în natură. În acest sens, conceptul de interacţiune este o categorie filozofică cu acelaşi grad de generalitate ca cel de mişcare. Precizez că între sistem şi mediul exterior se exercită ceea ce vom numi “interacţii externe” iar între diversele părţi componente ale sistemului se exercită ceea ce vom numi “interacţii interne”. Procesele din sisteme sunt o consecinţă a interacţiunilor externe plus a celor interne. Interacţiunile externe constau într-un schimb de energie şi de substanţă între sistem şi mediul înconjurator. Interacţiunile interne constau în schimb de energie şi de substanţă între diversele parţi ale sistemului (între subsisteme) dar mai pot consta şi în
4
schimbarea structurii (modificări de faza, de stare de agregare, stare chimică). Se cunosc în prezent patru tipuri de de interacţiuni elementare: - nucleare - electromagnetice - de dezintegrare beta - gravitaţia Pentru definirea procesului care este o consecinţă a interacţiunilor trebuie să se precizeze noţiunile de structura şi stare. 1.2.3. Starea sistemului termodinamic, parametri de stare Starea unui sistem termodinamic reprezintă totalitatea proprietăţilor lui la un moment dat. Proprietăţile care determină univoc starea sistemului termodinamic, în condiţiile fizice concrete în care se gaseşte acesta sunt caracterizate prin parametri de stare ai sistemului. Parametri de stare, mărimile care caracterizează diferitele proprietăţi ale sistemului se împart în : a) parametrii interni sau intensivi când nu depind de cantitatea de substanţă din sistem şi depind de natura şi modul de mişcare a constituienţilor, adică de ansamblul si distribuţia în spaţiu a acestor constituienţi; Presiunea, temperatura, densitatea, polarizarea electrica, sunt astfel de parametri. Aceşti parametri se mai numesc şi forţe generalizate. b) parametrii externi sau extensivi depind de mediul înconjurator cu care sistemul se gaseşte în interacţie şi de cantitatea de substanţă din sistem; sunt parametri aditivi ca de exemplu : volumul, acceleraţia gravitaţională, magnetizarea, masa. Aceşti parametri se mai numesc şi coordonate generalizate. Mai târziu vom arata ca sistemele termodinamice sunt caracterizate de proprietatea de ergodicitate, adică la echilibru parametrii interni sunt funcţii de parametrii externi şi de o alta variabilă care poate fi temperatura sau energia internă. Sistemul termodinamic în funcţie de posibilitaţile de interacţiune cu alte sisteme înconjuratore poate fi: • izolat dacă nu interacţionează cu mediul exterior a) izolat adiabatic: starea lui se modifica numai datorită modificării parametrilor externi b) izolat mecanic: starea sistemului se modifică fără variaţia parametrilor externi • închis daca schimbă energie dar nu substanţa cu mediul înconjurător • deschis dacă schimbă energie şi substanţă cu mediul înconjurator În raport cu structura sa, sistemul termodinamic poate fi: • omogen dacă are aceleaşi proprietăţi în toate punctele şi nu există interfeţe de
separare pentru părţile macroscopice ale sistemului cu componenţi diferiţi • neomogen (eterogen), dacă are proprietăţi diferite în diferite puncte ale sistemului. 1.2.4. Starea de echilibru termodinamic. Postulatul echilibrului sau principiul general al termodinamicii Dacă parametrii de stare ai unui sistem termodinamic nu se modifică în timp, starea se numeşte stare staţionară.
5
O stare stationară a unui sistem termodinamic este de echilibru dacă nu exista fluxuri, adică staţionaritatea stării sistemului nu este rezultatul unor interacţiuni externe în raport cu sistemul considerat (de exemplu nu primeşte şi nu cedează caldură, substanţă, etc.). Prin urmare echilibrul termodinamic implică atât staţionaritatea parametrilor sistemului cât şi stationaritatea condiţiilor exterioare sistemului. Acest mod de a defini echilibrul termodinamic este idealizat, întrucât în mod riguros, parametrii de stare ai sistemului mai prezintă încă mici variaţii în jurul valorilor medii la echilibru, variaţii care poarta numele de fluctuaţii. Studiul fluctuaţilor se face la fizica statistica. Studiul echilibrului termodinamic a condus la stabilirea a două postulate fundamentale ale termodinamicii, cunoscute sub numele de principiul general al termodinamicii şi respectiv principiul zero al termodinamicii. Pentru început vom vorbi despre primul postulat sau principiul general al termodinamicii. Constatările experimentale privind tendinţa de evoluţie spre echilibru a sistemelor termodinamice izolate a permis formularea principiului general al termodinamicii: un sistem termodinamic izolat ajunge întotdeauna dupa un interval de timp la echilibru şi nu poate ieşi de la sine din această stare. Caracteristicile acestui principiu general al termodinamicii sunt: a) are caracter director, aratând sensul evoluţiei sistemelor termodinamice şi exprimând ireversibilitatea lor; b) evidenţiază posibilităţile termodinamicii şi limitele acesteia cu privire la studiul proceselor naturale. Astfel, principiul general se aplică numai sistemelor macroscopice finite; extrapolarea sa la Universul infinit poate determina concluzii eronate (de exemplu tinderea universului privit ca sistem termodinamic la echilibru ar implica “moartea termică a Universului”) c) nu conţine precizari cu privire la intervalul de timp după care se atinge echilibrul. Descrierea atemporală a proceselor este o caracteristică şi o limitare importantă a termodinamicii proceselor reversibile. 1.2.5. Transformarea sau procesul termodinamic Orice schimbare a stării unui sistem termodinamic se numeşte transformare sau proces; aşadar, trecerea unui sistem termodinamic de la o stare la alta poarta numele de transformare sau proces. Transformarea sistemului este caracterizata de o serie de mărimi numite mărimi de proces care depind de stările prin care trece sistemul, deci de drumul urmat de sistem. Spre deosebire de acestea, mărimile de stare depind numai de starea sistemului la un moment dat, deci numai de parametrii stării considerate. În caracterizarea completă a evoluţiei sistemelor termodinamice intervin atât marimile de stare cât şi cele de transformare. Transformarea termodinamica, reprezentând modificari de stare ale sistemului trebuie raportate la o stare iniţială (σ i ) şi o stare finală (σ f ), trecerea de la starea iniţială la cea finală (σ i →σ f ) facându-se printr-o mulţime de stări intermediare, prin variaţia continua a parametrilor de stare. Variaţia parametrilor de stare determină modificarea corespunzătoare a mărimilor de stare şi de proces ale sistemelor conform relaţiei generale:
6
F dif
i
f
= ∫σ
σ
F
În cazul mărimilor de stare, variaţia dF dintre două stări arbitrare ale sistemului este independentă de drum pentru că aşa cum am spus, mărimile de stare sunt asociate unei anumite stări. Matematic, această proprietate se traduce prin faptul că variaţia dF reprezintă o diferenţială totală exactă şi atunci în transformarea σ i →σ f rezultă că:
dF F Fs s f s
i
f
σ
σ
σ σ∫ = −( ) ( i )
În cazul unui proces ciclic, această proprietate se scrie matematic astfel:
dFs∫ = 0
În cazul mărimilor de proces care depind de toate stările intermediare prin care trece sistemul, fiind astfel funcţionale asociate unei mulţimi de stari, variaţiile ale mărimilor de proces între două stări arbitrare ale sistemului vor depinde de drum şi variaţiile le vom scrie ca
dFp
Fp
δFp .
F Fif p
i
f
= ∫δσ
σ
Transformările pot fi cvasistatice sau necvasistatice (nestatice). Din experienţă se ştie că dacă un sistem aflat la echilibru este perturbat, sistemul revine la echilibru printr-un proces numit proces de relaxare. Se numeşte timp de relaxare şi se notează cu τ, timpul după care sistemul revine la starea de echilibru. El este o masură a vitezei de relaxare. Fie d t t dt tσ σ σ( ) ( ) ( ),= + − modificarea stării sistemului datorită variaţiei parametrilor de stare în intervalul de timp dt. Fie variabilele care caracterizează complet stările de echilibru ale sistemului termodinamic, cu:
x x x xk1 2, .... ... n
x x x xi iki
ni
1 2, ,.... .... valorile variabilelor în starea iniţială şi x x x xf f
kf
nf
1 2, .... .... valorile variabilelor în starea finală
şi ∆xk
τ viteza de stabilire a echilibrului.
Ca urmare, un proces se numeşte cvasistatic, dacă viteza de variaţie a parametrilor de stare este mult mai mică decât viteza de relaxare a sistemului:
dxdt
xk k<<∆τ
Dacă viteza de variaţie a parametrilor de stare ai sistemului este comparabilă cu viteza de relaxare, procesul se numeşte nestatic:
dxdt
xk k≤∆τ
Din analiza condiţiilor prezentate rezultă că în decursul unui proces cvasistatic, sistemul se găseşte permanent în stări de echilibru. Procesele cvasistatice sunt procese idealizate; sistemul trecând prin stari de echilibru infinit apropiate între ele, transformarea cvasistatica poate fi reprezentată printr-o curbă continuă.
7
8
σf
σi
x
X
σf
σiX
x
Transformarile reale nu sunt decât aproximativ cvasistatnefiind stari de echilibru. Procesele pot fi de asemenea procese reversibile sau ireversireprezentata prin: σ i →σ f şi σ i →σ i trecerea având loechilibru. Proprietatea fundamentală a proceselor cvasistaticeIreversibilitatea este o caracteristică a proceselor reale. Procesele reversibile nu sunt realizabile în natură, dar predeoarece, permit punerea în evidenţă a mărimilor de stare teprincipiilor termodinamice. Natura de “neechilibru “a proceselor ireversibile face careprezentate în diagrame. Aşadar, sistemul termodinamic poate fi caracterizatmărimi de transformare. Cea mai cunoscută marime sau funinternă iar lucrul mecanic şi căldura sunt funcţii de transform În termodinamica se vorbeşte despre funcţiile de trancantitativă a unei interacţiuni macroscopice a sistemului cu mAstfe, este energia transferată de la sistem la mediu sainteracţiunii termice, iar lucrul mecanic este energia schimmediul ca urmare a interacţiunii mecanice. Interacţiunea despre care vorbim este interacţiunea termodcare presupune interacţiuni de tip mecanic, termic etc., pentrfizic există interacţiune legată de alte concepte distincte. În consecinţă putem defini: a) înveliş adiabatic, sudecât interacţiune mecanică între sistem şi mediu şi b) învelpermite numai interacţiune termică între sistem şi mediul înco
ice, starile intermediare
bile. Reversibilitatea este c prin aceleaşi stări de
este reversibilitatea.
zintă o mare importanţă rmodinamice şi stabilirea
acestea să nu poată fi
prin mărimi de stare şi cţie de stare este energia are sau de proces. sformare ca fiind măsura ediul înconjurător. u invers, ca urmare a bată de către sistem cu
inamică (macroscopică), u că din punct de vedere
prafaţa care nu permite iş diaterm suprafaţa care njurator.
2.TEMPERATURA
Multe din mărimile macroscopice (volumul presiunea şi temperatura, de exemplu) sunt legate direct de percepţiile simţurilor noastre spre deosebire de proprietăţile microscopice dar pentru orice sistem mărimile macroscopice şi cele microscopice trebuie să fie legate între ele, deoarece ele nu sunt decât moduri diferite de descriere a aceleiaşi situaţii. Împletirea punctului de vedere microscopic cu cel macroscopic este o caracteristica a fizicii moderne. În particular primele pot fi exprimate cu ajutorul ultimelor. Astfel, presiunea unui gaz văzută macroscopic este măsurată cu un manometru. Privită microscopic, ea este legată de vitezele pătratice medii ale moleculelor care ciocnesc unitatea de suprafaţă şi care transferă impuls fluidului manometric. Analog, temperatura unui gaz măsurată cu termometrul poate fi legată de energia cinetică medie de translaţie a moleculelor. Începem examinarea fenomenelor termice cu un studiu al temperaturii. 2.1. Echilibrul termic Principiul zero al termodinamicii (sau al doilea postulat) este corelat cu alte proprietăţi ale echilibrului termodinamic, permiţând introducerea temperaturii empirice ca parametru de stare specific termodinamicii. Enunţul principiului zero rezultă din generalizarea unor concluzii rezultate din experienţă. Pentru început vom încerca să înţelegem sensul noţiunii de echilibru termic şi de temperatură.Vom considera pentru aceasta două sisteme termodinamice A şi B aflate fiecare dintre ele în mod independent în stare de echilibru caracterizate de presiunile şi volumele celor două sisteme.
Fig.2.1 Echilibrul termic între două sisteme A şi B despărţite printr-un perete diaterm.
Punem în contact aceste sisteme, astfel încât fiecare să poată acţiona asupra celuilalt, dar ambele izolate de mediul înconjurător. Peretele diaterm împiedică schimbul de masă dintre cele două sisteme şi orice interacţiune mecanică, electrică sau magnetică; în schimb lasă să treacă căldura. În momentul cuplării sistemului A cu B se constată că (A+B) nu este la echilibru dar tinde şi atinge starea de echilibru după un anumit timp conform principiului general al termodinamicii. Echilibrul care se stabileste în sistemul (A+B) are loc în urma “interacţiunii” dintre Aşi B prin peretele diaterm. Această interacţiune nu este nici datorită reacţiilor chimice, nici datorită transferului de masă, nici datorită interactiunii mecanice, nici datorită interacţiunii electrice; este o interacţiune de tip nou - interacţiune termică.
9
Această interacţiune este responsabilă pentru trecerea sistemului compus (A+B) către o stare de echilibru atât pentru A cât şi pentru B în comparaţie cu stările lor iniţiale. Interacţiunea termică se mai numeşte şi schimb de căldură, constituind un mod special de transmitere de energie între sistemele A şi B. Echilibrul care se stabileşte în sistemul (A+B) se numeşte echilibrul termic. Mărimile de stare ale ambelor sisteme aflate în echilibru termic nu mai sunt independente unele de altele, între ele existând o legătura funcţională: f(A,B)=0, f fiind de fapt o “măsură” a dezechilibrului sistemului AUB. Forma funcţiei depinde numai de natura celor două sisteme şi ar putea fi determinată printr-un număr mare de experimente, în care, pornind de la diverse stări iniţiale ale ambelor sisteme se observă atingerea echilibrului termic.
2.2. Tranzitivitatea echilibrul termic. Principiul zero al termodinamicii.
2.2.1. Tranzitivitatea echilibrul termic Să considerăm acum două sisteme A şi B separate printr-un perete adiabatic, fiecare dintre ele aflându-se în contact cu un al treilea sistem C, prin intermediul unor pereţi diatermici, întregul sistem fiind într-un înveliş adiabatic, la echilibru.
Fig.2.2. a) b) Deci între A şi B nu există schimb de căldură. Fiecare dintre sisteme sunt iniţial în echilibru.Între A şi C şi respectiv B şi C este permis schimbul de căldură (Fig.2.2 a). Experienţa arată că A şi B vor atinge echilibrul termic cu C şi nu va apare nici-o modificare a presiunii dacă peretele adiabatic dintre A şi B este înlocuit cu unul diaterm (Fig. 2.2.b). Sau, altfel spus, dacă admitem realizarea echilibrului termic pe rând între A şi C şi respectiv B cu C, atunci când A şi B se aduc în contact prin peretele diaterm se constată că de fapt ele sunt în echilibru termic între ele. Deci: două sisteme aflate în echilibru termic cu un al treilea, simultan sau succesiv, se află în echilibru termic şi între ele. Matematic putem scrie astfel: f(AUC)=0 f(BUC)=0 implică f(AUB)=0 Stările σ σA E C ( în echilibru), respectiv σ σB E C ( în echilibru), determină proprietăţile: • σ σC E A şi σ σC E B , relaţia de echilibru termodinamic este simetrică; • σ σA AE ,σ σB E B şi σ σC E C relaţia de echilibru termodinamic este reflexivă;
10
• dacă σ σA E C şi σ σB E C atunci σ σA E B relaţia de echilibru termic este tranzitivă. Proprietatea de tranzitivitate a echilibrului termic exprimă conţinutul principiului zero al termodinamicii sau tranzitivitatea este o proprietate generală a echilibrului termic dintre stări. Principiul zero a fost enunţat pentru prima oară de Maxwell (1891) şi s-a numit principiul zero din motive istorice, pentru că a apărut după ce se enunţase atât principiul I cât şi principiul al II-lea (enunţate prima oară de Clausius). Această discuţie exprimă ideea că sistemele aflate în aceste stări posedă o proprietate care asigură condiţia ca ele să fie în echilibru termic unele cu altele atunci când sunt puse în contact.Această proprietate se numeşte temperatură empirică. Temperatura unui sistem este o proprietate care determină dacă un sistem se află în echilibru termic cu alte sisteme, sau Temperatura empirică reprezintă un parametru care permite compararea stărilor aflate la echilibru termic. 2.2.2. Enunţuri echivalente ale principiului zero al termodinamicii • Temperatura empirică este un alt anunţ echivalent al principiului zero • O formulare mai formală dar poate mai fundamentală a principiului zero este: există o mărime scalară numită temperatură, care reprezintă o proprietate a tuturor sistemelor termodinamice (în stări de echilibru), astfel încât egalitatea temperaturilor este o condiţie necesară şi suficientă pentru echilibrul termic. Esenţa principiului zero este : există o mărime utilă numită “temperatură”. • Aşadar, principiul zero permite definirea temperaturii ca o mărime fizică măsurabilă, prin asocierea univocă a unei valori numerice, fiecărei stări de încălzire a corpurilor. Ca urmare a introducerii temperaturii ca mărime care caracterizează starea internă a sistemului termodinamic se poate formula un alt enunţ al principiului zero al termodinamicii: • la echilibru, starea unui sistem termodinamic este determinată de cei n parametrii externi şi de temperatură, σ θ= f x x xn( , ,... , )1 2 . Este evident că notiunea de temperatură este lipsită de sens pentru sisteme care nu se afla la echilibru termic. În general se admite că toţi parametri interni , la echilibru sunt funcţii de parametrii externi şi de temperatură, adică
X X X n1 2, ,....x x xn1 2, ,.....
X X x x xk k n= ( , ,.... , )1 2 θ 2.1 enunţ echivalent al principiului zero al termodinamicii. Acest enunţ al principiului zero este valabil pentru o mărime de stare foarte importantă, energia internă, U care la echilibru termodinamic se poate scrie:
U U x x xn= ( , ,....... , )1 2 θ 2.2 Sistemele termodinamice la echilibru termic supuse principiului zero al termodinamicii se numesc sisteme ergodice.Aşadar, principiul zero al termodinamicii arată caracterul ergodic al sistemelor termodinamice. O consecinţă foarte importantă a principiului zero al termodinamicii o constituie axioma inaccesibilităţii izoterme: pot exista stări oricât de apropiate de o stare dată, care nu pot fi atinse printr-o transformare izotermă (izotermele nu se intersectează).
11
Simţurile noastre pun în evidenţă proprietăţile corpurilor de a fi mai calde sau mai puţin calde şi permit stabilirea unei relaţii de ordine a acestor stări de încălzire. Aceste senzaţii dau numai informaţii calitative. 2.3.Măsurarea temperaturii. Scări de temperatură. Caracterizarea obiectivă şi cantitativă a stării de încălzire a corpurilor este posibilă, deoarece experienţa a arătat existenţa unor corelaţii între schimbarea stării de încălzire semnalată de simţuri şi modificarea unor proprietăţi fizice ale corpurilor, cum ar fi: dilatarea corpurilor, creşterea presiunii unui gaz (la volum constant), creşterea volumului (la presiune constantă), rezistenţa electrică. Constatarea corelaţiei între variaţia stării de încălzire şi dilatarea lichidelor, a condus încă din timpul Renaşterii la construcţia unor dispozitive (termometre), care au permis ca prin măsurarea variaţiei lungimii unei coloane de lichid să se determine variaţia stării de încalzire. Oricare dintre aceste proprietăţi poate fi folosită pentru construirea unui termometru adică pentru stabilirea unei scări particulare (empirice) de temperatură. Deci pentru a stabili o scara empirică de temperatură se alege o substanţă termometrică particulară şI o proprietate termometrica particulară a acestei substanţe.Apoi se defineste scara temperaturilor presupunând o relaţie monotonă continuă între proprietatea termometrică aleasă şI temperatura măsurată pe scara noastra particulară. De exemplu, substanţa termometrică poate fi un lichid într-un tub capilar de sticlă si proprietatea termometrică poate fi lungimea coloanei de lichid; sau substanţa termometrică poate fi un gaz închis într-un vas si tinut la volum constant iar proprietatea termometrica presiunea gazului si mai pot fi o mulţime de alte exemple. Este important însă să realizăm că fiecare alegere a substanţei şi a proprietăţii termometrice, împreuna cu relaţia admisă sau presupusă între proprietate şi temperatură, duce la o scară particulară (empirică) a temperaturilor ale căror indicaţii nu trebuie să concorde neapărat cu indicaţiile date de alte scări de temperatură definite independent. Să presupunem că am ales o substanţă termometrică şi reprezentăm prin X proprietatea termometrică pe care vrem s-o folosim în stabilirea unei scări de temperaturi. Funcţia θ( )X este funcţia termometrică iar forma ei determină scara de temperaturi. Măsurarea temperaturii presupune alegerea unei proprietăţi termometrice care să aibă o variaţie cu temperatura cât mai simplă să spunem liniară:
θ( )X =kX, 2.3 unde k este o constantă care trebuie evaluată. Prin alegerea formei liniare pentru funcţia termometrică,θ( )X , am fixat faptul că diferenţe de temperatură egale, sau intervale de temperatură egale, corespund la variaţii egale ale proprietăţii termometrice (X). Aceasta înseamnă, de exemplu, că ori de câte ori lungimea coloanei de mercur din termometrul de sticlă cu mercur variază cu o unitate, temperatura variază cu o cantitate fixă bine definită, indiferent de temperatura de pornire. Rezultă de asemenea că două temperaturi măsurate cu acelaşi termometru, se află în acelaşi raport ca şi valorile X corespunzătoare, adică:
12
θθ
( )( )
XX
XX
1
2
1
2
= 2.4
Pentru a determina constanta k şi deci pentru a etalona termometrul trebuie precizat un punct fix standard la care toate termometrele trebuie să indice aceeaşi temperatură θ . Se pot folosi de asemenea două repere fixe.
a) pentru un reper fix: θθ
( )( )XX
XX0 0
= şi ca urmare se determină
θ θ( )X XX
= 00
2.5
prin măsurarea proprietăţii termometrice X, θ 0 fiind fixat prin convenţie.
b) pentru două repere fixe: θθ( )( )XX
XX
1 1= şi θθ
( )( )XX
XX
2 = 2 ; se scad relaţiile şi se obţine:
θθ θ
=−−
XX X
2 1
2 1
2.6
Măsurarea temperaturii implică realizarea echilibrului termic între termometru şi sistem, fără a modifica esenţial temperatura sistemului. Exprimarea numerică a temperaturii pe baza legilor (2.5) sau (2.6) necesită stabilirea unei unităţi de temperatură prin considerarea reperelor termometrice corespunzătoare unor fenomene fizice reproductibile şi divizarea intervalului dintre repere în părţi identice echidistante numite grade de temperatură. Deci o stare uşor reproductibilă a unui sistem standard, ales convenabil se numeşte punct fix. Din 1954 (a 10-a Conferinţă Generală asupra Măsurilor şi Greutăţilor de la Paris) se foloseşte un singur punct fix, punctul triplu al apei, care reprezintă starea în care gheaţa, apa lichidă şi vaporii de apă coexistă în echilibru. Aceată stare poate fi obţinută numai la o anumită presiune şi este univoc determinată. Presiunea vaporilor de apă în punctul triplu este de 4,58 torr. Temperatura în acest punct fix standard este aleasă în mod arbitrar egală cu 273,16 K. Kelvinul este intervalul de temperatură egal cu unitatea. Dupa proprietatea termometrică utilizată pentru măsurarea temperaturii termometrele sunt de mai multe tipuri: Nr.crt.
Termometru
Proprietatea Relaţia T(X)
1 Gaz menţinut la volum constant
presiunea, p T K p
pptr
= 27316,
2 Gaz menţinut la presiune constantă
Volumul,V T K V
Vvtr
= 27316,
3 Rezistenţă electrică (la curent constant)
Rezistenţa electrică,R T K R
RRtr
= 27316,
4 Termocuplul (la presiune Tensiunea otoare,e T K e
eetr
= 27316,
termometrică
constantă si curent zero) termoelectrom
13
5 Lichid aflat într-un tub capilar
Lungimea L T K L
LLtr
= 27316,
Exemplu: Un termometru cu rezistenţă de platină are o rezistenţă 90,35 Ω atunci când el este introdus într-un vas pentru punctul triplu. Ce temperatură va arăta termometrul dacă este introdus într-un mediu pentru care rezistenţa sa devine 96,28 ? Ω
T K R KR = =27316 96 28
90 35280 6, ,
,,
Problema care se pune este dacă valoarea pe care o obţinem pentru temperatura unui sistem depinde de alegerea termometrului pe care-l folosim. Prin definiţie se asigură faptul că termometrele de diferite tipuri vor fi în concordanţă la punctul fix standard, dar ce se va întâmpla la alte puncte? În consecinţă, pentru a obţine o scară bine definită a temperaturilor, trebuie să alegem un tip particular de termometru ca termometru etalon sau standard. Alegerea va fi făcută nu pe baza convenţiei experimentale, ci prin cerinţa ca scara temperaturilor definită de de un termometru particular să se dovedească o mărime utilă în formularea legilor fizicii. Cele mai mici variaţii în citirile temperaturii se constată pentru diferitele termometre cu gaz la volum constant, ceea ce sugerează alegerea unui gaz ca substanţă termometrică standard. S-a constat că pe măsură ce cantitatea de gaz folosită într-un astfel de termometru şI deci presiunea sa este redusă, variaţiile indicaţiilor date de diferite termometre cu gaz (care folosesc gaze diferite) se reduc. Ca urmare este ceva fundamental în comportarea unui termometru cu gaz la volum constant şi presuni joase. Termometrul cu gaz la volum constant aşa cum este descris mai jos este termometrul care este folosit pentru a stabili scara temperaturilor folosita astăzi.
Fig.2.3. Un termometru cu gaz la volum constant. Atâta timp cât mercurul din stânga tubului manometric la acelaşi nivel pe scală (zero), volumul gazului închis va fi constant. Meniscul poate adus la zero prin ridicarea sau coborârea rezervorului R.
14
Termometrul cu gaz la volum constant corpul termometric fiind gazul (de exemplu heliu) foloseşte presiunea la volum constant ca proprietate termometrică. Termometrul este prezentat schematic în fig. 2.3.El constă dintr-un balon de sticlă, porţelan, cuarţ, platină sau platină-iridiu (depinde de intervalul de temperatura pentru care este folosit) legat printr-un tub capilar de un manometru cu mercur. Balonul care conţine gazul este introdus într-o baie sau în mediul în care trebuie măsurată temperatura. Când temperatura gazului creşte, gazul se destinde, determinând coborârea mercurului în ramura B şI urcarea în A. Tuburile A şI B comunică printr-un tub de cauciuc cu rezervorul de mercur R. Ridicând sau coborând rezervorul cu mercur, mercurul din ramura B poate fi făcut să coincidă cu un reper fix (E), păstrând astfel tot timpul gazul la volum constant. Întrucât volumul se păstrează constant, starea sistemului este caracterizată numai de presiunea p care se determină uşor citind diferenţa de înălţime dintre coloanele de mercur A şi B şi cunoscând presiunea atmosferică: p p g= +0 hρ . Folsind relaţia din tabel se determină temperatura mediului. 2.3.2. Scări de temperatură După modul de alegere a fenomenelor fizice particulare şi dupa valorile numerice atribuite temperaturilor reperelor fixe se cunosc mai multe scări termometrice: Celsius, Fahrenheit. Reamur şi scara termodinamică. Scara termodinamică absolută a temperaturilor, numită şi scara Kelvin (Kelvin a propus-o pe baza principiului al doilea al termodinamicii) este o scara independentă de proprietăţile oricărei substanţe particulare. Scara gazului ideal si scara Kelvin sunt identice în intervalul de temperatură în care poate fi folosit termometrul cu gaz şi din acest motiv scriem K pentru unitatea de temperatură dată de termometrul cu gaz. cele mai folosite scări de temperatură sunt scara Celsius şi scara Fahrenheit. Ele se definesc cu ajutorul scării Kelvin. Scara Celsius a temperaturii foloseşte un grad (unitatea de temperatură) care are aceeaşi mărime ca şi unitaea Kelvin. ( Această scară a fost inventată de suedezul Celsius în 1742, a fost numită scară centigrad până în 1948 când la cea de-a 9 Conferinţă a Măsurilor şi Greutăţilor a fost numită scara Celsius). Relaţia de legătură dintre temperatura în K şi cea în grade Celsius este: T(K)=t ( 0C ) + 273,16 K Cele doua repere pentru scara Celsius au fost temperatura la care apa îngheaţă la presiune atmosferică normală 0 0C , care corespunde pe scara Kelvin temperaturii de 273,15 K şi temperatura la care vaporii de apă şi apa lichidă sunt la echilibru la presiunea atmosferică normală, 100 0C . Relaţiile dintre temperaturi pentru diferitele scări termometrice se găsesc în tabelul următor. Denumirea scării
Scara Celsius ( 0C )
Scara Rankine ( 0 Ra )
Scara Fahrenheit 0 F
Scara Reamur ( 0 R )
Celsius - 59
273150T Ra − , t F0 321 8−,
1 25 0, t R
Rankine
- t F0 459 67+ , 18 1 25 273150, ( , ,t R +
18 273160, ( , )t C +
15
Fahrenheit 1 8 320, t C + T Ra0 459 65− , - 94
320t R +
Reamur 0 8 0, t C 0 8 5
927310, ( ,T Ra −
49
320( )t F − -
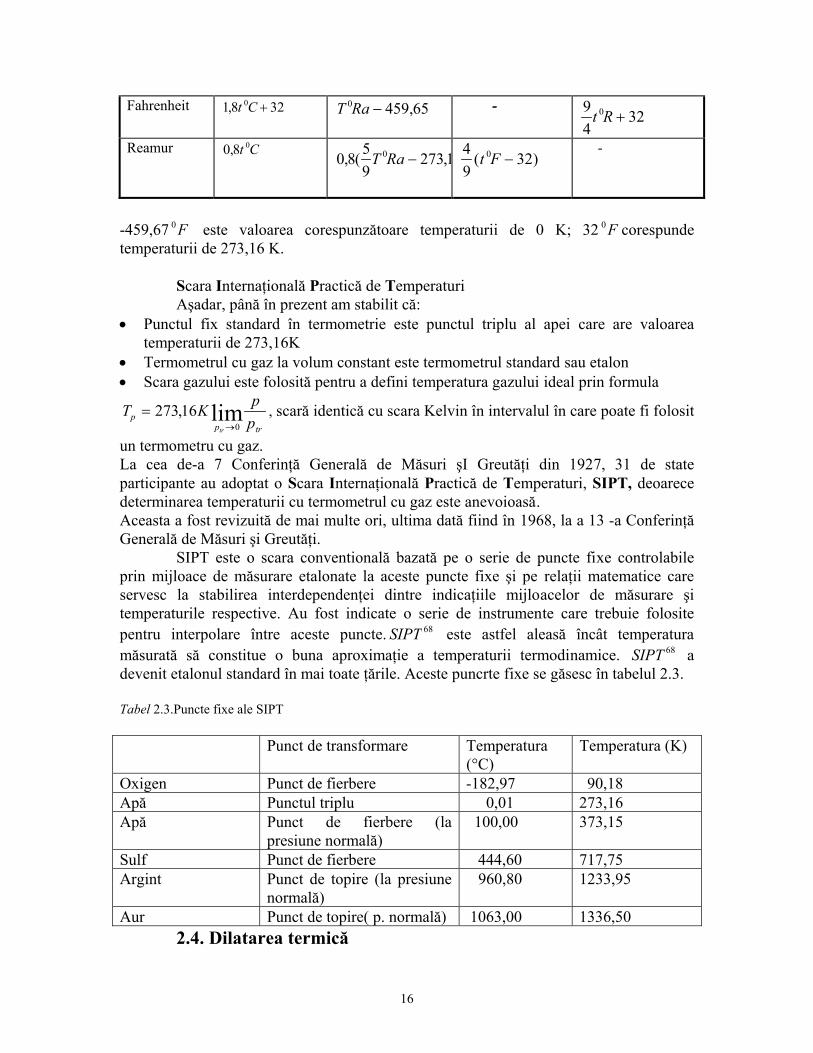
-459,67 0 este valoarea corespunzătoare temperaturii de 0 K; 32 corespunde temperaturii de 273,16 K.
F 0 F
Scara Internaţională Practică de Temperaturi Aşadar, până în prezent am stabilit că: • Punctul fix standard în termometrie este punctul triplu al apei care are valoarea
temperaturii de 273,16K • Termometrul cu gaz la volum constant este termometrul standard sau etalon • Scara gazului este folosită pentru a defini temperatura gazului ideal prin formula
T K ppp
p trtr
=→
273160
, lim , scară identică cu scara Kelvin în intervalul în care poate fi folosit
un termometru cu gaz. La cea de-a 7 Conferinţă Generală de Măsuri şI Greutăţi din 1927, 31 de state participante au adoptat o Scara Internaţională Practică de Temperaturi, SIPT, deoarece determinarea temperaturii cu termometrul cu gaz este anevoioasă. Aceasta a fost revizuită de mai multe ori, ultima dată fiind în 1968, la a 13 -a Conferinţă Generală de Măsuri şi Greutăţi. SIPT este o scara conventională bazată pe o serie de puncte fixe controlabile prin mijloace de măsurare etalonate la aceste puncte fixe şi pe relaţii matematice care servesc la stabilirea interdependenţei dintre indicaţiile mijloacelor de măsurare şi temperaturile respective. Au fost indicate o serie de instrumente care trebuie folosite pentru interpolare între aceste puncte. SIPT 68 este astfel aleasă încât temperatura măsurată să constitue o buna aproximaţie a temperaturii termodinamice. SIPT 68 a devenit etalonul standard în mai toate ţările. Aceste puncrte fixe se găsesc în tabelul 2.3. Tabel 2.3.Puncte fixe ale SIPT Punct de transformare Temperatura
(°C) Temperatura (K)
Oxigen Punct de fierbere -182,97 90,18 Apă Punctul triplu 0,01 273,16 Apă Punct de fierbere (la
presiune normală) 100,00 373,15
Sulf Punct de fierbere 444,60 717,75 Argint Punct de topire (la presiune
normală) 960,80 1233,95
Aur Punct de topire( p. normală) 1063,00 1336,50 2.4. Dilatarea termică
16
Efectele obişnuite ale variaţiilor de temperatură sunt variaţiile în dimensiunea şi starea materialelor. Vom considera variaţiile dimensiunilor care se produc fără modificarea stării substanţei. Variaţia dimensiunii unui corp solid cum este lungimea sa, lăţimea sau grosimea , se numeşte dilatare liniară. Dacă lungimea acestei dimensiuni liniare este l, variaţia lungimii, determinată de variaţia T∆ a temperaturii este .l∆ Din experienţă s-a constatat că, dacă T∆ este suficient de mic, variaţia .l∆ a lungimii este proporţională cu variaţia de temperatură T∆ şi cu lungimea iniţială l. Prin urmare se poate scrie:
Tll ∆⋅⋅=∆ α 2.7 unde α , numit coeficient de dilatare liniară, are valori diferite pentru materiale diferite. Ecuaţia (2.7) se poate rescrie pentru a obţine expresia pentru α :
Tl
l ∆∆
=1α [ ] 11
.−− == KgradISα
α are semnificaţia variaţiei relative a lungimii în raport cu variaţia temperaturii cu un grad. Valoarea lui α depinde de temperatura la care se determină lungimea l, dar variaţia lui α este de obicei neglijabilă în raport cu precizia cu care se efectuează măsurătorile.Ca urmare se consideră constantă pentru un material dat. În tabelul 2.4. sunt date valorile experimentale ale coeficientului mediu de dilatare liniară pentru mai multe substanţe. Tabel 2.4. Valori ale coeficientului de dilatare liniară, α Substanţa α , grad-1 Substanţa α , grad-1 Aluminiu 23⋅10-6 Cauciuc tare 80⋅10-6
Pentru multe solide, numite izotrope, variaţia procentuală (relativă) a lungimii pentru o variaţie dată de temperatură este aceeaşi pentru toate liniile din solid. Dilatarea este analoagă unei măriri fotografice, cu excepţia faptului că solidul este tridimensional. Dacă
o placă plană are o gaură în ea, )( Tll
∆=∆ α pentru o variaţie T∆ este acelaşi pentru
lungimea, grosimea, diagonala feţei, diagonala spaţială şi diametrul găurii. Orice linie, fie dreaptă fie curbă, se lungeşte în raportul α pentru un creşterea cu un grad a temperaturii. Cu o foarte mare precizie variaţia relativă a ariei A pentru un variaţia temperaturii cu un grad, în cazul unui solid izotrop, este 2α , adică
TAA ∆⋅⋅=∆ α2 iar variaţia relativă a volumului V pentru o variaţie cu un grad a temperaturii pentru pentru un solid izotrop, este 3α , adică:
17
TVV ∆⋅⋅=∆ α3 Deoarece forma unui fluid nu este definită, are sens numai variaţia volumului cu temperatura. Gazele răspund rapid la variaţii de temperatură sau presiune, în timp ce variaţia de volum a lichidelor cu variaţia temperaturii sau presiunii este foarte mică. Se
notează cu β coeficientul de dilatare volumică a unui lichid, TV
V ∆∆
=1β şi s-a dovedit că
acest coeficient de dilatare volumică este independent de temperatură. Lichidele se dilată cu creşterea temperaturii, dilatarea volumică fiind de aproximativ zece ori mai mare decât a solidelor. Apa, cel mai obişnuit lichid, nu se comportă ca celelalte lichide. În figura 2.4.este prezentată curba dilatării apei.La valori mai mari decât 4°C apa se dilată odată cu creşterea temperaturii, deşi neliniar. La valori între 4°C şi 0°C, însă, apa se dilată în loc să se contracte. Dilatare la scăderea temperaturii nu se observă la niciun alt lichid.;se mai observa la substanţe de tipul cauciucului si la anumite substanţe solide cristaline pe intervale limitate de temperatură. Densitatea apei este este maximă la 3,98°C când are valoarea aproximativă de 1000 kg/m3.La celelalte temperaturi densitatea apei este mai mică. Aceasta comportare a apei explică de ce lacurile îngheaţă mai întâi la suprafaţă. Fig.2.4.Variaţia densităţii apei cu temperatura la presiunea atmosferică normală (a);variatia densităţii intre 0°C şi 10°C (b). ÎNTREBĂRI ŞI PROBLEME 1. Ce fel de concept este temperatura ,microscopic sau macroscopic? 2. Este vreun gaz mai bun decât altul pentru folosirea într-un termometru etalon cu gaz la volum constant? Care sunt proprietăţile gazului necesare pentru un astfel de termometru? 3. Se poate folosi un termometru de sticlă cu apă? De ce este mai bun termometru de sticlă cu mercur?
18
4. Dacă temperatura în scara gazului la punctul de fierbere al apei este 373,15 K, care este valoarea limită a raportului dintre presiunea unui gaz la punctul de fierbere al apei şI presiunea la punctul triplu al apei, dacă gazul este menţinut la volum constant? 5. Fie p presiunea din balonul unui termometru cu gaz la volum constant, atunci când balonul se afla la temperatura punctului triplu de 273,16 K şI fie p presiunea atunci când se află la temperatura camerei. Fie date trei termometre cu gaz la volum constant:
tr
1) gazul este oxigenul şI =20 cm Hg; ptr
2) gazul este oxigenul şI =40 cm Hg; ptr
3) gazul este hidrogenul şi =30 cm Hg. ptr
Valorile p măsurate pentru aceste trei termometre sunt: p1, p2, p3. a) O valoare aproximativă a temperaturii camerei, T, poate fi obţinută cu fiecare din termometre folosind formulele:
T Kp
cmHg1127316
20= , ; T K
pcmHg2
22731640
= , ; T Kp
cmHg3327316
30= ,
Marcaţi cu “adevărat” sau “fals” fiecare din următoarele afirmaţii: (1) Cu ajutorul metodei descrise toate cele trei termometre vor da aceeaşi valoare; (2) Cele doua termometre cu oxigen vor fi în concordanţă unul cu celălat dar nu cu termometrul cu hidrogen; (3) Fiecare din cele trei termometre va da o valoare diferită pentru T. b) În cazul în care există o discordanţă între cele trei termometre, să se explice cum ar trebui modificată metoda de utilizare a acestora pentru ca toate cele trei termometre să dea aceeaşi valoarte pentr T. 6. La ce temperatură scara Fahrenheit şI scara Celsius ar indica aceeaşi temperatură? Dar scara Fahrenheit şI scara Kelvin? 7. Temperatura suprafeţei Soarelui este de aproximativ 6000 K. Să se exprime această temperatură în scara Fahrenheit. 8.Să se exprime temperatura normala a corpului uman 98,6 în scara Celsius. 0 F 9. Un termometru greşit etalonat introdus într-un amestec de apă cu gheaţă indică -8 grade, iar când este în apă care fierbe el indică +112 grade. Experienţa se realizează la presiune atmosferică normală. Să se determine temperatura reală când termometrul arată +40.
19
3. PRINCIPIUL ÎNTÎI AL TERMODINAMICII (L,Q,U,H)
Scimbarea stărilor de echilibru ale sistemului termodinamic, deci efectuarea de către acesta a procesului termodinamic se poate produce numai ca urmare a interacţiei dintre sistemul considerat şi mediul înconjurător.
3.1 Principiul echivalenţei dintre lucrul mecanic şi căldură
Dintre cele trei principii ale termodinamicii, principiul întâi ca legea transformării şi conservării energiei este cel mai general. Faptul că această lege se aplică la toate sistemele din natură, indiferent de “domeniul tradiţional” al acestora, a constituit o cucerire epocală pentru gândirea umană. Legea generala a conservării şi transformării energiei a fost stabilită pe trei căi diferite de trei savanti eminenţi: R. Mayer, J. Joule şi Helmholtz. Această lege a apărut mai întâi ca o lege care lega două seturi de mărimi şi două unităţi din ştiinţe considerate până atunci complet diferite: mecanica şi calorimetria sau electricitatea şi calorimetria. Este vorba pe de o parte lucrul mecanic (L) sau lucrul mecanic electric şi energia mecanică (cinetică şi potenţială) cu unităţile lor de măsură iar pe de altă parte căldura (Q) şi ulterior energia internă (U) cu unitatea de măsura caloria sau kcal. Pe baza observaţiilor sale asupra unor fenomene naturale diferite medicul şi fizicianul R.Mayer a formulat pentru prima dată (1842) legea transformării şi conservării energiei sub forma cea mai generală. A pornit de la a presupune echivalenţa dintre L şi Q şi a calculat raportul acestor mărimi, determinând pentru acesta, pe baza datelor experimentale, o valoare constantă. Clausius a formulat (1850) principiul întâi fără să considere căldura iar Poincaree a folosit experienţa lui Joule de echivalenţă cantitativă între lucru mecanic şi căldură şi a introdus energia internă. Planck a postulat matematic principiul întâi iar Caratheodory foloseşte noţiunea suplimentară de înveliş adiabatic care permite să se facă distincţie între diversele forme de energie. 3.1.1. Echivalenţa dintre L şi Q. Experienţa Joule. Între anii 1844 şi 1854 fizicianul englez Joule a efectuat experienţe care au marcat profund dezvoltarea ştiinţei. El îşi propusese să stabilească o relaţie între lucrul mecanic consumat pentru degajarea de căldură şI căldura degajată. Trebuie sa precizez că până la acest moment calorimetria se dezvoltase si avea metode suficiente de determinare a căldurii. Fig. 3.1.
h
1 2 3
G
S 1. vas de cupru izolat termic şi plin cu apă; 2.agitator înzestrat cu palete; 3. palete fixate pe pereţii vasului, care să frâneze mişcarea apei în timpul rotaţiei agitatorului.
18
Agitatorul este pus în mişcare de rotaţie de o greutate (G) prin intermediul unui cablu trecut peste un scripete (S). Prin caderea greutăţii pe o distanţă h, lucrul mecanic efectuat de greutate (deci şi de agitator) este egal cu scăderea energiei potenţiale cu semn schimbat, adică cu Gh. Pentru a calcula caldura degajată în vasul plin cu apă se măsoară cu un termometru temperatura apei. Joule a stabilit ca există o proportionalitate între lucrul mecanic cheltuit L şi căldura Q obţinută: Q= A⋅L; J = L/Q. « A » reprezintă un coeficient de proporţionalitate, acelaşi întotdeauna, indiferent de modul de obţinere a căldurii, de forma de lucru mecanic, de temperatura corpurilor. Cu alte cuvinte, Joule a stabilit că acelaşi lucru mecanic produce totdeauna aceeaşi căldură. Aşadar s-a demonstrat: căldura obţinută era echivalentă cu lucrul mecanic consumat; această echivalenţă rămâne adevărată chiar dacă L este produs prin consum de căldură. R. Mayer a calculat valoarea J a raportului dintre L şi Q. Experienţa lui Joule poate fi interpretată în perspectiva ştiinţei actuale, astfel: Dacă după caderea greutăţii G s-a înregistrat o creştere a temperaturii în calorimetru, ∆t, (presupunând că s-a pornit de la o temperatură t initială egală cu temperatura mediului), aceasta s-a obţinut pe seamă efectuării de lucru mecanic. Apoi, îndepărtând învelişul adiabatic şi punând calorimetrul în contact termic cu mediul înconjurător, între acestea existând diferenţa de temperatură ∆t, apare o interacţiune termică manifestată printr-un schimb de căldură Q, până când se restabileşte echilibrul termic calorimetru - mediul înconjurător. În acest moment sistemul (calorimetrul) a revenit la starea iniţială. Se spune că sistemul a parcurs un ciclu termodinamic format din două procese: a) a primit lucru mecanic în condiţii adiabatice ;b) a cedat căldura Q mediului ambiant şi a revenit la starea initială.
t∆
t0
t0
Q
Cum în experienţa lui Joule s-au folosit mai multe căderi succesive ale greutăţii G înseamnă că L este de fapt o sumă, respectiv la limită o integrală a lucrului mecanic elementar, δL, extinsă pe întreg ciclul, δL∫ .
Această sumă (integrală) va fi egala conform cu rezultatul experimentului lui Joule, întotdeauna cu cu suma căldurilor elementare, δQ cedată de sistem mediului ambiant, daca sistemul revine la starea iniţială, bineînţeles înmulţită cu echivalentul mecanic al caloriei pentru ca egalitatea să fie omogenă dimensional:
δ δL J= ∫∫ Q 3.2
sau dacă folosim echivalentul caloric al lucrului mecanic, A=1/J: A L Qδ δ= ∫∫ 3.3
Această relaţie reprezintă transcrierea matematică a principiului echivalenţei dintre căldură şi lucru mecanic, aparţinând lui Poincaree (1888).
19
Acest rezultat fiind generalizat pentru orice sistem termodinamic care poate interacţiona mecanic (δL) şi termic (δQ) cu mediul exterior, parcurgând un ciclu, conduce conform cu Poincaree-Joule la introducerea noţiunii de energie internă. Ecuaţia 3.3 se scrie:
( )δ δQ A L− =∫ 0 3.4
şi folosind proprietatea integralelor ciclice care spune că dacă integrala ciclică dintr-o mărime este zero, atunci mărimea integrată este o diferenţială totală exactă a unei marimi de stare, rezultă că:
δ δQ A L dU− = 3.5 Considerând A≡1 (aceleaşi unităţi de măsură pentru Q şi L).
dU= 3.6 δQ - δL relaţie care defineşte noţiunea de energie internă a sistemului termodinamic.Deci experienţa lui Joule conduce la introducerea unei mărimi de stare energia internă,U. (noua în raport cu mecanica, termometria si calorimetria). 3.2. Lucrul mecanic Aşa cum am mai spus una din formele de interacţiune dintre sistemul termodinamic şi mediul exterior este lucrul mecanic. Din mecanică se stie ca acesta apare datorită existenţei unor forţe care se exercită între sistem şi exterior. Conform principiului acţiunii şi reacţiunii, forţele cu care sistemul acţioneaza asupra mediului sunt egale în mărime dar de sens contrar celor cu care mediul exterior acţionează asupra sistemului. Noi vom înţelege prin forţe de interacţie, forţele exercitate de sistem asupra mediului înconjurător. Alţii folosesc convenţia invers. Sub acţiunea forţelor, parametrii externi ai sistemului variază şi sistemul în urma transformării trece de la o stare la alta. În mecanică, lucrul mecanic elementar δL efectuat de o forţă când punctul său de aplicaţie se deplasează cu
rF
drr este:
cu i=x,y,z 3.7 δL Fdr F dri ii
= ==∑
r r
1
3
Ca urmare lucrul mecanic într-o transformare finită de la o stare iniţială σ i la o stare finală σ f va fi:
3.8 L Fi iii
f
==∑∫
1
3
σ
σ
dr
Pentru calculul integralei este necesar sa se cunoască nu numai starea iniţiala şi finală ci şi toate stările intermediare, integrala depinzând de drum. Câmpurile de forţe pentru care integrala nu depinde de drum ci numai de starea initială şi cea finală se numesc câmpuri de forţe conservative. În cazul unui sistem termodinamic cu n parametrii externi ,ai fiecărui parametru extern i se poate asocia o forţă generalizată ,Ai având în conformitate cu principiul zero al termodinamicii forma generală:
A A cu i=1,2,....n 3.9 a a a Ti i n= ( , ..... , )1 2
Ai fiind parametrii interni ai sistemului. Ai sunt parametrii conjugaţi parametrilor externi a . i
În acest caz lucrul mecanic elementar efectuat de sistem caracterizează modificarea stării sistemului termodinamic datorită parametrilor externi sub acţiunea forţelor generalizate asociate (conjugate):
20
δ a L A di ii
==∑
1
3
3.10
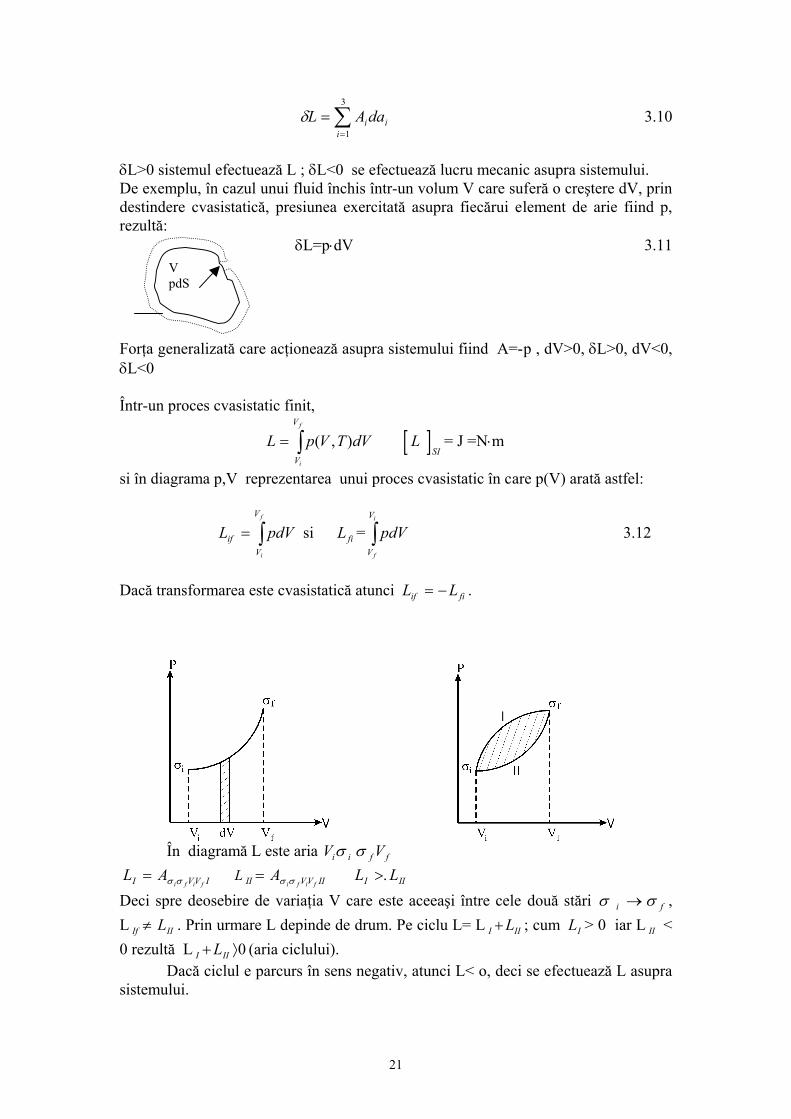
δL>0 sistemul efectuează L ; δL<0 se efectuează lucru mecanic asupra sistemului. De exemplu, în cazul unui fluid închis într-un volum V care suferă o creştere dV, prin destindere cvasistatică, presiunea exercitată asupra fiecărui element de arie fiind p, rezultă:
δ
V
L=p⋅dV 3.11 V pdS
Forţa generalizată care acţionează asupra sistemului fiind A=-p , dV>0, δL>0, dV<0, δL<0 Într-un proces cvasistatic finit,
L p V T dV
V
i
f
= ∫ ( , ) [ ]LSI
= J =N⋅m
si în diagrama p,V reprezentarea unui proces cvasistatic în care p(V) arată astfel:
si = 3.12 L pdVifV
V
i
f
= ∫ Lfi pdVV
V
f
i
∫ Dacă transformarea este cvasistatică atunci L Lif fi= − .
În diagramă L este aria Vi iσ σ f fV
L AI i f i f= σ σ V V I L II V V IIA
i f i f= σ σ L LI I>. I
Deci spre deosebire de variaţia V care este aceeaşi între cele două stări σ i → σ f , L . Prin urmare L depinde de drum. Pe ciclu L= LIf IIL≠ I IL I+ ; cum > 0 iar L < 0 rezultă L (aria ciclului).
LI II
I IIL+ ⟩0 Dacă ciclul e parcurs în sens negativ, atunci L< o, deci se efectuează L asupra sistemului.
21
∫ pdV ≠ 0 3.13
Lucrul mecanic efectuat de sistem într-un proces nestatic este întotdeauna mai mic decât L în cvasistatic. (de demonstrat,vezi exerciţiul) Exemple de lucru mecanic: 1 Lucrul mecanic de alungire a unei bare.Considerăm o bară elastică de lungime l, având un capăt fixat; la celălalt capăt se exercită o forţă de tracţiune F (în cazul în care F este negativ, tracţiunea devine compresiune). Lucrul mecanic elementar la o alungire dl este: δL=Fdl.
0
2 mecanic de torsiune.Considerăm aceeaşi bară fixată la un capăt şI presupunem că la celălalt capăt se exercită un cuplu de torsiune.
0 Lucrul
La o răsucire cu unghiul dθ cele două puncte de aplicare ale forţelor se deplasează cu r dθ. Lucrul mecanic elementar al celor două forţe este:
δL=2rF dθ Dar 2rF este tocmai momentul M al cuplului, deci putem scrie: δL =M⋅ dθ
Fr
3 mecanic al tensiunii superficiale la variaţia suprafeţei.Se ştie că, dacă se consideră ca sistem o porţiune din suprafaţa unui fluid, porţiunile înconjurătoare exercită asupra porţiunii considerate o forţă care, raportată la unitatea de lungime a curbei care mărgineşte porţiunea considerată, se numeşte tensiune superficială şi se notează cu σ. Aceasta forţă este tangentă la suprafaţă, normală la elementul de arc al curbei care mărgineşte sistemul şi îndreptată spre exteriorul sistemului.
0 Lucrul
Folosind acelaşi raţionament ca la lucrul mecanic al forţei de presiune dar ţinând seama de diferenţa de sens între presiune si tensiunea superficială se obţine pentru lucrul mecanic al acesteia la o creştere (algebrică) cu ds a ariei suprafeţei, expresia: δL= σds, unde σ este tensiunea superficială. S-ar mai putea da exemple de lucru mecanic datorită electrizării sau magnetizării corpurilor. De reţinut că dacă un sistem este supus simultan mai multor feluri de solicitări, lucrul mecanic elementar este suma lucrurilor efectuate de fiecare solicitare în parte.
3.3.Principiul întâi al termodinamicii pentru procesele adiabatice. Energia internă
Orice transformare care se petrece într-un sistem izolat într-un înveliş adiabatic se numeşte transformare adiabatică. Fie un sistem termodinamic închis într-un vas prevazut cu piston ca în figură. Sistemul în întregime este înconjurat de un înveliş adiabatic. Din starea initială a sistemului în starea finală se poate trece astfel: a) la căderea greutăţii se transmite lucru mecanic L1, prin intermediul paletelor
moriştii;
22
b) L1se măsoară prin înalţimea pe care cade greutatea G; sistemul îşi modifică
volumul prin deplasarea pistonului;L2 se calculează ştiind forţa şi deplasarea pistonului.
c) atât prin deplasarea pistonului cât si a greutăţii, L3. Experienţa arată ca pentru aceste transformări L1= L2=L3
)
, aşadar L nu depinde de drum când procesul este adiabatic. Rezultă atunci că exista o functie U, proprietate a sistemului care depinde de starea sistemului, astfel încât lucrul mecanic într-o transformare de la σi→σf este:
L pdV U Uif f ii
f
= = − −∫ (σ
σ
3.14.
Funcţia U se numeşte energia internă a sistemului iar Uf şi respectiv Ui reprezintă valorile ei în starea finală respectiv iniţială. Lucrul mecanic se efectuează pe seama scăderii energiei interne ( de aici semnul minus). Deci, într-o transformare a unui sistem adiabatic:
∆U= Uf - Ui = -L 3.15 Enuţul de bază (primar) al principiului întâi (concluzia rezultatelor experimentale): Daca un sistem este închis într-un înveliş adiabatic, atunci lucrul mecanic efectuat de sistem într-o transformare oarecare depinde numai de starea initială şi starea finala a sistemului (forţele sunt conservative). Relaţia 3.15. defineste energia internă şi dă posibilitatea măsurării ei, pentru ca lucrul mecanic este măsurabil. Astfel, U este o funcţie univoca de stare şi deci dU este o diferenţiala totală exactă.U este definită până la o constanta aditivă arbitrară. Pentru a preciza valoarea acestei constante, se alege în mod arbitrar o anumita stare ca stare de referinţă căreia i se atribuie în mod convenţional valoarea zero: Uf= U0 si rezultă Ui = L, adica energia internă a oricarei stări este numeric egala cu L efectuat de sistemul izolat adiabatic pentru a ajunge din starea respectivă în starea de referinţă. Sau Ui= U0 şi Uf = - L. Dacă transformarea este ciclică, σi =σf, Uf=Ui şi L=0. Sub forma diferenţială, principiul întâi pentru formularea adiabatică se scrie:
dU = -δL 3.16. U, energia interna astfel definita se referă la energia tuturor formelor de mişcare şi de interacţie dintre particulele care alcătuiesc sistemul. Dacă la energia internă a sistemului se adaugă energia sa cinetică ( de mişcare în ansamblu a sistemului) şi cea potenţială datorită prezenţei unui câmp de forţe extern, se obţine energia totală a sistemului. În studiul nostru, al legilor mişcării termice, se poate considera numai energia internă a sistemului, alegându-se în mod corespunzător ipotezele de lucru.
23
3.4.Căldură. Forma generală a principiului întâi al termodinamicii.
3.4.1. Căldura Noţiunea de căldură joacă un rol cu totul deosebit în termodinamică. Urmărind momentele importante din istoria fizicii, noţiunea de căldură este strâns legată de evoluţia a două concepte fundamentale diferite de tratare a fenomenelor termice. Unul este cel al continuumului, iar cealălt este cel al discontinuităţii materiei. Pe de o parte t.c.m.(teoria cinetico moleculară) a oferit o explicaţie intuitivă pentru acest concept şi a unor fenomene asociate cu acesta, dar nu a putut duce la un mijloc de măsurare a ei. În schimb metoda fenomenologică, prin conceptul (abandonat de altfel ) al caloricului a condus la calorimetrie şi deci la o tehnică experimentală foarte precisă de măsurare a căldurii. Orice noţiune din ştiinţele exacte ale naturii capătă o recunoaştere unanimă abia în momentul în care devine fie direct măsurabilă cu un aparat fie este calculata cu ajutorul unei relaţii în care intră mărimi măsurabile. Căldura face parte din a doua categorie, adică ea nu este direct măsurabilă, dar calculul ei se bazează pe măsurători directe ale unor mărimi:m(kg), t (0C). Căldura este măsura schimbului de mişcare termică între corpuri. Căldura masoară deci energia schimbată între corpuri pe calea interaţiunii termice. Interactiunea termică cunoaşte trei forme: conducţia, convecţia si radiaţia. Când spunem cantitatea de căldură schimbată între două corpuri înţelegem energia schimbată între acele corpuri pe calea unuia, a două sau a tuturor celor trei tipuri de interacţiune termică. Conducţia- interacţiunea termică care se manifestă la contactul dintre două corpuri aflate la temperaturi diferite.Temperaturi diferite înseamnă la scară moleculară dezechilibru energetic, adică moleculele unui corp au o stare de agitaţie mai intensă decât ale celuilalt corp. Convecţia- interacţiunea termică care apare la interfaţa dintre un fluid în mişcare în raport cu un corp solid aflat la o temperatură diferită de cea a fluidului. Radiaţia- interacţiunea termică dintre două corpuri de temperaturi diferite aflate la distanţă unele de altele, prin intermediul radiaţiei termice. Fie un sistem izolat adiabatic ca în fig,3.4: A
AQA<0
A
B
QA >0 0
Fig.3.4. A este sistemul, B este mediul înconjurător
. (UAf+UBf) - (UAi + UBi ) = -L = 0 ( nu există variaţie a parametrilor externi) Ca urmare, ∆UA = -∆UB Căldura schimbată de A cu mediul exterior (B) reprezintă variaţia energiei interne a mediului B. Deci schimbul de căldură s-a făcut pe seama scăderii energiei interne a sistemului B:
Q UB 3.17. A = - ∆Se face urmatoarea convenţie: QA >0, sistemul primeşte căldură Q ul cedează căldură A <0, sistem
24
Sistemele A şi B în contact termic şi izolate adiabatic de exterior ca în fig.3.4 implică , ţinând seama de ec 3.17 că : QA + QB= 0 , sau căldura primită de unul din sisteme este egală cu cea cedată de celălalt sistem.
Ca urmare, pentru mai multe sisteme izolate putem scrie: = 0 Qii
n
=∑
1
3.4.2. Formularea generală a principiului întâi Considerăm sistemele A şi B ca în fig.3.5.
B AA
Fig.3.5. A-sistem termodinamic B-mediul exterior Se efectuează L asupra sistemului A. Ca urmare:
(UAf+ UBf) - (UAi - UBi) = - L 3.18. sau (UAf - UAi) + (UBf - UBi) = -L ; UAf - Uai = - (UBf - UBi) - L Deci, pentru sistemul A ţinând seama de ec 3.17 rezultă:
UAf - Uai = QA - L 3.19 Ca urmare, pentru orice alt sistem care nu este izolat adiabatic, în cursul unei transformări, L depinde de drum şi :
U 3.20 f - Ui = Q - L Ecuatia 3.20 reprezintă formularea generală a principiului întâi al termodinamicii (Q primit, L efectuat). Pentru un proces infinitezimal ec 3.20 se scrie:
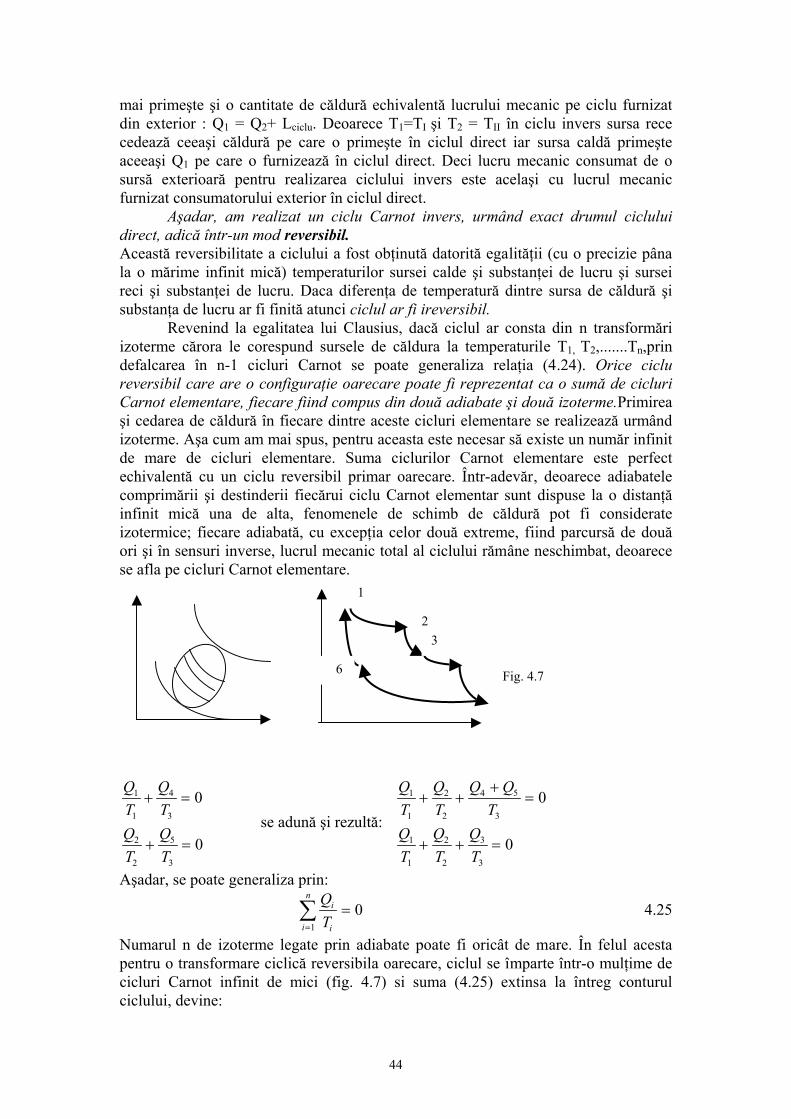
dU = δQ - δL 3.21. Ecuatia 3.21 constituie formularea matematică generală a principiului întâi al termodinamicii sub forma diferenţială şi se enunţă astfel: Variaţia energiei interne a unui sistem termodinamic care evoluează între două stări date este determinată de lucrul mecanic efectuat de către sistem (sau asupra sistemului) şi căldura schimbată de sistem în cursul procesului. Sau formulat o serie de enunţuri pentru principiul întâi cu referire la fenomenele termice: a) Pentru un ciclu:
dU Q L= − =∫∫ ∫δ δ 0 3.22
Ecuaţia 3.22 exprima echivalenţa dintre lucru mecanic şi căldură descoperită experimental de Joule. b) nu se poate realiza o maşina termică care să produca lucru mecanic fără să primească căldură.Imposibilitatea construirii unui perpetuum mobile de speţa întâi. c) Energia interna a unui sistem izolat este constantă: Uf - Ui = U (Q = L =0), sau legea conservarii energiei. În concluzie: • Principiul întâi este aplicabil în toate procesele fie ele cvasistatice sau
necvasistatice;are caracter de generalitate. • introduce energia internă ca funcţie de stare; • nu indică sensul de desfăşurare a proceselor.
25
3.5. Proprietatile termice caracteristice unui sistem termodinamic Utilizându-se principiile zero şi întâi ale termodinamicii se pot pune în evidenţă atât proprietăţi ale sistemelor termodinamice corelate cu temperatura sistemului cât şi proprietăţi determinate de căldura implicată în procesele studiate. Astfel de proprietăţi pot fi studiate utilizându-se ecuaţiile termice de stare şi ecuaţia calorică de stare care constituie împreună un sistem de n+1 ecuaţii corespunzătoare celor n+1 grade de libertate ale sistemului termodinamic, adică de numarul de parametri independenţi care caracterizează starea sistemului. 3.5.1. Ecuaţiile termice de stare Ecuaţiile corespund exprimării forţelor generalizate ale sistemului termodinamic funcţie de parametrii externi şi temperatură pe baza principiului zero al termodinamicii, sub forma:
A 1, a2, ......an; T) 3.23 i = Ai (aEcuaţia calorică de stare U=U(a1,a2,......an;T) 3.23’
Ec 3.23. reprezintă ecuaţia termică de stare; denumirea este condiţionată de faptul că prin intermediul acestor ecuaţii se calculează temperatura. Ecuaţiile termice de stare pot fi privite ca modele matematice (uneori deduse din experienţă, alteori din modele fizice). Exemple: a) Ecuatia termică de stare a gazelor perfecte a rezultat fie din experienţă (Clapeyron - Mendeleev) pe baza legilor simple determinate anterior (Boyle - Mariotte, Gay-Lussac) sau din teoria cinetico moleculară (Clausius) pentru un model fizic. Pentru un sistem termodinamic simplu există numai doi parametrii independenţi şi astfel A = A(a,T). Gazul ideal este un astfel de sistem şi A = p, a = V şi deci p = p(V,T), sau f(p,V,T) = 0 reprezintă ecuaţia termică de stare. Ecuaţia pV= νRT este totodată un model explicativ (intuitiv), contribuind la explicarea semnificaţiei fizice a presiunii, temperaturii, energiei interne. b) Fluidul Van der Waals Ecuaţia termică de stare a gazelor perfecte nu explică particularităţile de comportare a gazelor reale şi nu prinde fenomenul modificării stării de agregare. Pentru gazele reale s-au stabilit numeroase ecuaţii de stare (peste 150) a căror exprimare este cu atât mai complicată cu cât aproximează mai bine comportarea reală a gazelor. Între aceste ecuaţii un rol deosebit l-a jucat ecuaţia caracteristică stabilită de Van der Waals (1873), folosind considerente cinetico-moleculare.
( )p aV
V b RT+
− =
02 0 3.24
( )p aV
V b R+
− =
ν ν ν2
2 T
unde a şi b sunt constante: a-datorita forţelor de atractie dintre molecule; b- datorită forţelor de respingere dintre molecule. Ecuatia Berthelot (1903):
( )p aTV
V b RT+
− =
02 0 3.25
26
Această ecuaţie a fost folosită pentru evaluarea constantei R a gazelor perfecte şi pentru determinarea coeficienţilor termici. Ecuaţia virială de stare Dezvoltarea virială este dezvoltarea presiunii unui sistem (de obicei fluid) în
serie Mac Laurain, după densitate. De obicei se scoate în factor termenul V
mRTµ
pentru compararea cu ecuaţia gazului ideal:
....))()(1( 2
2
+++= TCVmTB
VmRT
Vmp
µ 3.26
B şi C sunt funcţii numerice de T şi se numesc coeficienţi viriali. Deşi la început a fost considerată ca o ecuaţie empirică, treptat ecuaţia virială de stare a ajuns să prezinte o importanţă deosebită, fiind apreciată astăzi ca unica ecuaţie de stare care dispune de o bază teoretică solidă. Aceşti coeficienţi se calculează în fizica statistică, plecând de la potenţialul de interacţie reciprocă dintre molecule. Deci, prin comparaţie cu datele experimentale se pot face evaluări pentru forţele intermoleculare. Pentru gazul ideal clasic, B(T)=0 şi C(T)=0.
În general, pentru un gaz, mV
B T( ) este termenul cel mai relevant al dezvoltării
viriale şi într-o primă aproximaţie putem scrie:
))(1( TBVmRT
Vmp +=
µ
La temperaturi joase, energia potenţială de atracţie dintre molecule (negativă) este mult mai mare decât energia cinetică (pozitivă). Ca urmare, presiunea exercitată de molecule asupra pereţilor vasului este diminuată de atracţia reciprocă a moleculelor şi deci B(T) trebuie să fie negativ. Dimpotrivă, dacă gazul se încălzeşte, energia cinetică devine mai mare decât cea potenţială şi presiunea începe să crească: B(T) devine pozitiv.
. B(
T
Fig. 3.6. Variaţia lui B(T) măsurată pentru heliu în stare gazoasă
Din cele spuse se poate trage concluzia că modelul gaz ideal este un caz particular de dezvoltare virială; la fel modelul gazului van der Waals. 3.5.2. Coeficienţii termici Întotdeauna când există nişte cauze care determină transformări ale sistemului termodinamic există şi un răspuns al sistemului; funcţiile de răspuns sunt variaţii ale mărimilor de stare (volum, presiune) când unul sau mai multi parametrii sunt modificaţi în mod controlat. Ele sunt singurele mărimi accesibile masurătorilor şi singurele mijloace de a construi ecuaţii de stare pentru un sistem, pornind de la experienţă.
27
10 Coeficienţi de dilatare termică(p=ct) - mărime fizică care măsoară variaţia relativă a volumului când temperatura variază cu o unitate, menţinându-se presiune constantă:
α = ⋅+ −1
0 0
0 0 0 0
V p TV p T T V p T
T( , )( , ) ( , )∆
∆ ca rezultat al măsurătorilor experimentale.
α ∂∂α=
→
=∆T
pVVT
0
1lim [ ]α S I grd. . = −1 3.27
20 Coeficientul termic al presiunii - marimea fizică care măsoară variaţia relativă a presiunii când temperatura variază cu un grad, volumul menţinându-se constant.
β = ⋅+ −1
0 0
0 0 0 0
p V Tp V T T p V T
T( , )( , ) ( , )∆
∆, măsurat experimental şi deci:
β ∂∂
β=
→
=∆T
VppT
0
1lim [ ]β S I grd. . = −1 3.28
30 Coeficientul de compresibilitate (compresiune) - marimea fizică care măsoară variaţia relativă a volumului când presiunea variază cu o unitate la T =const.→ coeficient de compresiune izotermă S = const. coeficient de compresibilitate adiabatică →
kV p T
V p p T V p TpT = − ⋅
+ −1
0 0
0 0 0
( , )( , ) ( ,∆
∆0 )
k TVpT
pT
kV
=
→
=−∆ 0
1lim ∂∂
[ ]k mNT S I. . =
2
3.29
kV p T
V p p S V p SpS = − ⋅
+ −1
0 0
0 0 0
( , )( , ) ( ,∆
∆0 )
k SVpS
pS
kV
=
→
=−∆ 0
1lim ∂∂
[ ]k mNS S I. . =
2
3.30
4 0 Relaţii importante între coeficienţii termiciPentru orice sistem termodinamic (indiferent de ecuaţiile de stare) există o relaţiegenerală de legătură între coeficienţii termici: pentru f(x, y, z) = o
∂∂
∂∂
∂∂
xy
yz
zzxz x y
= −1 3.31
numită relaţie de ciclicitate
28
Dacă aplicăm relaţia de ciclicitate sistemului termodinamic simplu caracterizat de parametrii p,T, şi V se obţine:
∂∂
∂∂
∂∂
VT
Tp
zpVp V T
= −1 3.32
Din această relaţie se obţine legătura dintre coeficienţii termici studiaţi: α β= pkT 3.33
Aceasta relaţie este foarte importantă în practică pentru determinarea lui β în cazul corpurilor solide şi lichide întrucât este imposibil să încălzim corpul fără variaţia volumului. În plus, cunoaşterea coeficienţilor termici implică cunoaşterea ecuaţiei termice de stare. Demonstraţia ecuaţiei de ciclicitate: Fie ecuaţia termică de stare: p=p(V,T), pe care o diferenţiem
dp pV
dV pT
dTT V
=
+
∂∂
∂∂
Se consideră presiunea constantă şi se împarte ecuaţia cu dT, rezultând:
∂∂
∂∂
∂∂
pV
VT
pTT p V
+
= 0 sau ∂∂
∂∂
∂∂
pV
VT
pTT p
= −
V
care evident
devine relaţia de ciclicitate:
∂∂
∂∂
∂∂
pV
VT
TpT p V
= −1
a) Dacă se cunoaşte ecuaţia termică de stare se pot determina coeficienţii termici. Exemplu gazul ideal: pV=νRT
V RTp
=ν şi ∂
∂νV
TRpp
= deci pentru că α ∂∂
=
1V
VT p
atunci α =1T
p RTV
=ν şi ∂
∂νp
TR
VV
= β ∂∂
=
1p
pT p
β =1T
∂∂
νVp
RTpT
= − 2 k
VVpT
T
=
1 ∂∂
kpT =1
pVγ=const. S
S pV
Vk
=
∂∂1
pS γ1
=k
b) Invers, dacă se cunosc coeficienţii termici de stare se poate determina ecuatia de stare.
De exemplu dacă: α =1T
şi kpT =1γ
se poate deduce ecuaţia de stare V=V(T,P)
dV VT
dT Vp
dpp T
=
+
∂∂
∂∂
şi dV VdT Vk dpT= +α şi ca urmare: dVV
dTT
dpp
= −
ln ln ln lnV T p− + = c şi deci pV/T =const. 3.6 Proprietăţile calorice ale sistemelor termodinamice
29
Procesele în care este implicată căldura, variaţia ei în cazul când parametrii interni sau externi pot să rămână constanţi determină proprietăţile calorice ale sistemului termodinamic. Ecuaţia calorică de stare U = U(a1,....an,T) arată variaţia energiei interne datorită temperaturii şi condiţiilor mecanice ( prin parametrii externi). Pentru determinarea proprietăţilor calorice este necesar, în general să se cunoască nu numai ecuaţia calorică dar şi ecuaţiile termice. Dintre proprietăţile calorice fac parte înainte de toate, capacităţile calorice si căldurile latente. Aceste mărimi pot fi determinate experimental şi astfel din principiul I. putem exprima variaţia de energie în funcţie de ele, pentru că trebuie spus că expresia ecuaţiei calorice de stare (şi a ecuaţiei termice) nu se pot determina teoretic decât prin fizica statistică. 3. 6. 1. Proprietăţile calorice ale sistemelor termodinamice la parametri
externi constanţi
Conform principiului I, δQ se datoreşte atât variaţiei de temperatură cât şi variaţiei parametrilor externi ai sistemului (ai). Astfel:
δQ=dU(ai,T) + ( )A T a daii
i i∑ , 3.34
δQ= ∂∂
∂∂
UT
dT Ua
A daa i T
ii
ii
+
+
∑ 3.35
dacă considerăm un sistem termodinamic simplu, adică un sistem cu 2 parametrii ca de exemplu: a = V şi A = p
δQ= ∂∂
∂∂
UT
dT UV
p dVV Ti
+
+
3.36
Ecuaţiile 3.35 şi 3.36 pot fi folosite în definirea constantelor calorice ale sistemului, astfel: - capacităţiile calorice la parametrii externi constanţi: reprezintă căldura schimbată de sistem cu mediul exterior la o variaţie a temperaturii sistemului egală cu unitatea, când parametrii externi se păstrează constanţi:
Cai = iadT
Q
δ capacitatea calorică [ ]C J
Ka S Ii .=
cai = iadT
Qm
δ1 3.37
căldura specifică[ ]c JkgKS I. =
CiadT
Q
δυ
1 caldura molară [ ]C J
kmol KS I. =⋅
ai =
-călduri latente: reprezintă caldurile schimbate de sistem cu exteriorul pentru o variaţie a param. externi ai sistemului cu unitatea când temperatura se păstrează constantă şi aj≠ai constanţi. Căldurile latente indică posibilitatea sistemului de a schimba energie cu mediul exterior în mod izoterm prin variaţia parametrilor externi.
30
ji
i
aaTia da
Q
≠
=
,
δλ şi ji
i
aaTia da
Q
≠
=Λ
,
1 δν
3.38
Ţinând seama de ecuaţiile 3.35 şi 3.36 şi de definiţii putem să exprimăm coeficienţii calorici în funcţie de energia internă şi de parametrii externi.
C UTa
ai
i
=
∂∂
şi λ ∂∂a
i Tii
Ua
A=
+ 3.39
şi ca urmare ecuaţia calorică de stare 3.35 se scrie: δQ C dT daai
= + λ a ii∑ 3.40
care reprezintă o forma Pfaff de ordinul întâi, lineară în T şi parametrii externi. Pentru un sistem termodinamic simplu, pentru care p=p(V,T) coeficienţii calorici sunt:
C UVV
V
=
1ν
∂∂
ΛVT
UV
p=
+
1ν
∂∂
3.41
şi ca urmare ecuaţia calorică de stare 3.36 se scrie : δ ν νQ C dT dVV V= + Λ 3.42
3.6.2. Proprietăţile calorice în cazul parametrilor interni constanţi - ENTALPIA Dacă într-o transformare cvasistatică se menţin constanţi parametrii interni Ai (forţele generalizate) asociaţi parametrilor externi ai , din principiul I scris sub forma:
δQ=dU(ai,T) + ( )A T a daii
i i∑ , 3.43
prin integrare, când sistemul evoluează între stările σ1 şi σ2 se obţine:
3.44 δσ
σ
σ
σ
Q U U A daii
i
1
2
1
2
2 1∫ ∑ ∫= − +
δ 1σ
σ
Q U A a U A a H Hi ii
ii
i
1
2
22
11
2∑ ∑= + − + = −( ) ( )∫ 3.45
Deci Q depinde numai de starea iniţială şi finală. Mărimea H, definită prin relaţia H U A ai
ii= + ∑ 3.46
poartă numele de entalpie, reprezintă o proprietate a sistemului şi este o funcţie de stare şi are o importanţă deosebită în studiul proceselor termodinamice. Dacă sistemul termodinamic este simplu:
H= U + pV 3.48 şi δQ= dH reprezintă semnificaţia fizică a entalpiei: entalpia este o mărime de stare a cărei variaţie într-un proces izobar ne dă cantitatea de căldură schimbată de sistem cu mediul exterior în timpul interacţiei termice. Pentru evidenţierea proprietăţilor calorice în cazul menţinerii parametrilor interni constanţi, vom scrie ecuaţia calorică cu ajutorul entalpiei: H(A i ii, T) = U + A a dH dU a T d A ai
ii i∑ ∑⇒ = +( , ) ( )
A da a dA+∑ ∑
sau: dH dU a Ti ii
i ii
i= +( , )
şi conform principiului întâi:
31
dH A T Q a dAii
i( , ) = + i∑δ 3.49.
şi pentru sistem termodinamic simplu:
δ
δ
Q dH A T a dA
Q dH Vdp
i ii
i= −
= −
∑( , ) 3.50
Mărimile calorice la parametrii interni constanţi vor fi: • Capacităţile calorice la parametrii interni constanţi: căldura schimbată de sistem cu mediul exterior la o variatie a temperaturii cu o unitate şi parametrii interni constanţi; • Caldurile latente la parametrii interni constanţi care arata posibilitatea sistemului de a schimba căldură cu mediul exterior, când parametrii interni sunt constanţi. Capacităţile calorice:
C δ
capacitatea calorică QdT Ai
[ ]C J
KA S Ii .= Ai =
ciAdT
Qm
δ1 căldura specifică[ ]c J
kgKS I. = 3.51 Ai=
C 1υ
caldura molară [ ]δQdT Ai
C Jkmol KS I. =
⋅ Ai =
şi căldurile latente:
ji
i
AATiA dA
Q
≠
=
,
δλ Λ Ai T A A
i
i j
QdA
=
≠
1ν
δ
,
3.51’
Ecuaţiile (3.50) şi (3.50’) devin, dezvoltând entalpia ca funcţie de stare:
δ ∂∂
∂∂
Q HT
dT HA
a dAA i T
ii
ii
=
+
−
∑
δ ∂∂
∂∂
Q HT
dT Hp
V dpp T
=
+
−
Din definiţiile coeficienţilor calorici rezultă expresiile lor ţinând seama de expresiile ecuaţiei calorice generale şi pentru sistem termodinamic simplu:
C HTA
Ai
i
=
∂∂
λ ∂∂A
i Tii
HA
a=
−
şi o altă exprimare pentru ecuaţiile calorice: δ λQ C dT dAA A
ii i
= + ∑ i
C HTp
p
=
1ν
∂∂
Λ pT
Hp
V=
−
1ν
∂∂
şi respectiv:
δ ν νQ C dT dpp p= + Λ Aşadar, când variabilele independente sunt Ai şi T în locul energiei interne se foloseşte ca funcţie de stare entalpia.
32
3.6.3.Relaţii generale între coeficienţii termici şi coeficienţi calorici • Cp- CV=?
δQ = ∂∂
∂∂
UT
dT UV
p dVV Ti
+
+
C QdTp
p
=
1ν
δ şi ca urmare:
C 1 1
ν
∂∂ ν
∂∂
∂∂
UT
UV
p VTV Ti
+
+
p =
p
Dar C =1 U
VVV
ν
∂∂
ΛVT
UV
p=
+
1ν
∂∂
iar ∂∂
αVT
Vp
=
C αΛVV p- CV = Aşadar, pentru determinarea diferenţei dintre cele două călduri molare este nevoie să se cunoască şi ecuaţia termică de stare. • Relaţia dintre Cp şi Cv pe de o parte şi kT şi kS pe de altă parte.
C HTp
p
=
1ν
∂∂
C UVV
V
=
1ν
∂∂
cu Cp> CV
δ ν νQ C dT dpp p= + Λ δ ν νQ C dT dVV V= + Λ
Considerând procesele adiabatice se obţine:SV
p
S
S
V
p
V
p
VkTVTp
CC 1
Λ
Λ−=
Λ
Λ=
∂∂∂∂
Se consideră ca sistemul schimbă aceeaşi căldură în procese izoterme: ν νC dT dpp p+ =Λ ν νC dT dVV V+ Λ cu dT = 0 Ca urmare :
Λ
ΛV T
Vp
∂∂
p = şi deci:
STV
p
VkpV
CC 1
=
∂∂
Relaţia dintre coeficienţii termici şi calorici este:
S
T
V
p
kk
CC
=
33
4.PRINCIPIUL AL II -LEA AL TERMODINAMICII Istoria acestui principiu este una dintre fascinantele aventuri ale ştiinţei, care a
generat nenumărate paradoxuri, controverse şi predicţii tulburătoare (moartea termică), presărată cu evenimente uneori tragice (sinuciderea lui Boltzmann), o aventură care a atras irezistibil o serie de minţi geniale ale omenirii, revoluţionari dintre cei mai mari ai fizicii (Planck, Einstein), nenumăraţi laureaţi ai premiului Nobel.
4.1.Caracterizarea generală şi conţinutul principiului al doilea al termodinamicii
Această pasionantă aventură a început cu prima revoluţie tehnico-ştiinţifică (crearea maşinii cu abur şi preocupările legate de îmbunătăţirea randamentului acesteia - Carnot), generând cercetări ce s-au desfăşurat pe un fundal din ce în ce mai larg si mai profund, culminând în anii secolului XX cu o contribuţie extrem de importantă la actuala revoluţie tehnico-ştiinţifică (cibernetică, informatică), odată cu introducerea conceptului de entropie informaţională (Shannon) prin analogie cu entropia statistică Boltzmann. Esenţa principiului al doilea constă în introducerea unei noi mărimi de stare entropia şi în precizarea sensului de variaţie a acesteia în sistemele izolate ( Afanasieva 1928). Principiul al II-lea indică sensul în care se desfăşoară procesele din natură, stabileşte limita maximă de transformare a căldurii în lucru mecanic în procese ciclice si afirmă neechivalenţa calitativă dintre L si Q. Primul principiu al termodinamicii a arătat posibilitatea transformării L în Q si invers fără a specifică în ce condiţii aceste transformări sunt posibile.El a arătat echivalenţa cantitativă dintre L si Q si a introdus proprietatea de energie internă (U), care nu variază în absenţa acţiunilor exterioare pentru orice procese din interiorul sistemelor. Din definiţia noţiunilor de L si Q s-a constatat că între acestea există o deosebire fundamentală: dacă lucrul mecanic poate determina variaţia oricărei forme de energie, căldura poate determina numai variaţia energiei interne a sistemului. Această neechivalenţă dintre Q si L nu ar avea importanţă dacă s-ar putea transforma fără dificultate Q în L. După cum arată experienta, în timp ce la transformarea L în Q fenomenul se poate limita la variaţia stării termodinamice numai a corpului (receptorul de căldură, de exemplu la încălzirea prin frecare sau electrică), la transformarea Q în L, odată cu răcirea corpului care cedează Q, are loc variaţia stării termodinamice a altor corpuri care iau parte la proces: a substanţei de lucru la procesele deschise sau a altor corpuri la procesele ciclice închise. Variaţia stării corpului de lucru (dacă procesul este deschis) sau cedarea unei părti din caldură de către substanţa de lucru altor corpuri şi în consecinţă schimbarea stării termodinamice a acestor corpuri într-un proces ciclic de transformare a Q în L se numeşte compensaţie. Experienţa conduce deci la faptul că fără compensaţie nu se poate transforma nici -o calorie de căldură în lucru mecanic. Dar lucrul mecanic se poate transforma în căldură fără compensaţie. Cu alte cuvinte, dacă căldură este transformastă în lucru mecanic şi după un proces ciclic de la un corp sau de la diferite corpuri s-a luat o cantitate de căldură Q (Q<0) iar L efectuat este L (L>0):
34
Q > L (sageata indică sensul procesului) Daca dimpotrivă, L (L<0) este transformat în caldură, atunci L = Q Deci principiul al II-lea al termodinamicii reprezintă ansamblul a doua propoziţii independente:
Q > L 4.1
L = Q
care exprimă sub formă cantitativă conţinutul principiului al II-lea al termodinamicii. Prin urmare, mişcarea ordonată (L) poate fi transformată integral în mişcare dezordonată (Q), pe când procesul invers este IMPOSIBIL, necesitând un mecanism compensator auxiliar.
4.2. Transformarea monotermă.Formularea de bază a principiului al doilea
Conform cu principiul I, pentru un sistem care suferă o transformare ciclică, ∆U=0 şi Q=L. În consecinţă există situaţiile: a) L = 0; Q = 0
b) L > 0; Q > 0 4.2 c) L < 0; Q < 0 Când este realizat cazul (b) se spune că sistemul funcţionează ca o maşină termică: primeşte căldură şi efectuează lucru mecanic. Problema cu largi aplicaţii practice este cum să se transforme Q în L cu randament maxim. Pentru aceasta este necesar în primul rând un rezervor termic (termostat), adică un corp sau un sistem de corpuri care se găsesc în stare de echilibru termodinamic şi care au o rezervă suficientă de energie internă sau altfel spus, o capacitate calorică infinită. Rezervorul nu are posibilitatea să efectueze el însuşi lucru mecanic ci doar să schimbe energie sub forma de căldură cu alte corpuri. Sistemul care primind energie internă sub formă de căldură, de la unul sau mai multe rezervoare termice şi efectuează L se numeşte corp de lucru (substanţă de lucru). Dispozitivul care cu ajutorul unui corp de lucru transformă periodic (continuu) Q în L se numeşte masină termică.
Prin căderea greutăţii G pe distanţa h, L se transformă în căldură. Căldura Q încălzeşte R(θ) mărindu-i energia internă.
Fig.4.2. Reprezentarea instalaţiei de conversie a L şi Q şi încălzirea unui rezervor termic. Întrebare de principiu: Este posibil ca M, corpul de lucru,
R(θ)
Q L M
35
primind caldura Q de la termostatul R(θ) să o transforme integral în L pe seama căruia G să fie ridicat la înălţimea h iar corpul de lucru să revină la starea initială, gată să transforme o noua cantitate de căldură în L fără alte schimbări în mediu? Cu alte cuvinte: Este posibil ca o masină termică a cărui corp (substanţă) de lucru, schimbând Q cu un singur termostat sa efectueze o transformare ciclică monotermă făra schimbări în mediul exterior?
O astfel de maşină se numeşte maşină monotermă şi schema ei este dată în figura 4.3.
R(θ)
Q L M
Fig.4.3. Schema maşinii monoterme
Carnot a fost primul care a arătat că nu este posibil să funcţioneze o maşină cu un singur rezervor (termostat), având nevoie de cel puţin două. În natură nu exista un proces ciclic monoterm, deci nu se poate realiza conversia periodică a lui Q în L fără modificări în mediul exterior, afirmaţia inversă fiind valabilă (Fig.4.2.) Aşadar, într-o transformare ciclica monotermă este imposibil să se transforme căldura în lucru mecanic făra schimbări în mediul exterior. Conţinutul acestui postulat este cunoscut sub denumirea de “formularea de bază a principiului al doilea al termodinamicii” Instalaţia care ar transforma periodic, fară compensaţie căldura unui corp oarecare în lucru mecanic se numeşte perpetuum mobile de speţa a II-a. ( W.Ostwald 1853-1932). Formularea de bază se regăseşte sub două forme, cele ale lui Clausius şi Thomson: 1 W. Thomson (Lord Kelvin): nu este posibil un proces ciclic reversibil în cursul caruia sa fie transformată în L, Q primită de la o singură sursa de
0 Postulatul
căldură şi nu se poate construi un perpetuum mobile de speţa a II-a. 2 a Clausius: Căldura nu trece de la sine de la un corp cu o temperatură dată la un corp cu o temperatură mai ridicată.
0Axiom
Se poate demonstra că cele două formulări sunt echivalente.
4.3. Fapte experimentale care au condus la principiul al doilea al termodinamicii. Procese reversibile si ireversibile Cel de-al II-lea principiu al termodinamicii arată prin experienţă că nu pot
exista procese în care să aibă loc transformarea căldurii în lucru mecanic fără compensaţie (transformarea necompensată a Q în L) şi în al doile rând posibilitatea existenţei altor procese care nu sunt legate de o asemenea transformare. Astfel, principiul al doilea duce la împărţirea proceselor în reversibile şi ireversibile (exp. din fig.4.1- proces ireversibil). Procesul de trecere a unui sistem termodinamic din starea σ1 în starea σ2 se numeşte reversibil dacă trecerea σ2→σ1nu este legată de o transformare necompensată a Q în L; procesul de trecere a unui sistem termodinamic din starea σ1 în starea σ2se numeste ireversibil dacă trecerea σ2→σ1 este legată de o transformare necompensată a Q în L.
36
Evident, orice proces cvasistatic este reversibil, Experienţa arată că, exceptând două procese nestatice cunoscute până acum, supraconductibilitatea şi suprafluiditatea, toate procesele nestatice (reale) sunt ireversibile. Exemple de procese ireversibile: 10 Destinderea unui gaz în vid.
Fig. 4.4. Experienţa lui Joule privind detenta adiabatică a gazului perfect în vid. Sistemul după deblocarea pistonului ocupa
întreg volumul şi pentru că în destinderea adiabatică în vid pext =0,δL = 0, δQ = 0 care implică dU = 0 şi ca urmare proces izoterm ∆T = 0. Readucerea sistemului în starea initială (principiul I nu interzice acest lucru) trebuie realizată prin comprimarea cu consum de L din exterior. Dar prin comprimare gazul se încălzeşte şi căldura respectivă trebuie convertită integral în lucru mecanic consumat, ceea ce nu este realizabil fără modificări în mediul exterior, deci făra compensaţie.
Vid gaz
20 Procesul de difuzie în gaze Acest proces este de asemenea ireversibil. Într-adevăr, dacă într-un balon se află doua gaze separate printr-un perete si se scoate apoi peretele, fiecare gaz va difuza în celalalt. Pentru a le separa trebuie ca fiecare gaz să fie comprimat; în acest caz pentru ca gazele să nu se încălzească este necesar să se ia căldură de la ele iar pentru ca această caldură să nu producă modificări în mediul exterior ar trebui ca să se transforme în lucru mecanic fără compensaţie, ceea ce nu este posibil. 3 de transmitere a căldurii la o diferenţă de temperatură finită este ireversibil, întrucât trecerea inversă este legată de luarea unei cantităţi determinate de căldură de la corpul rece, de transformare în lucru mecanic şi de cheltuirea ei pentru creşterea energiei corpului încălzit. Ireversibilitatea rezultă şi din faptul că el este nestatic.
0 Procesul
40 Procesul de frecare În procesul de frecare, lucrul mecanic poate fi transformat în căldură fără compensaţie, întrucât trecerea inversă a sistemului din starea finală în cea iniţială este legată de transformarea necompensată a căldurii în lucru mecanic; rezultă ca procesele în care există frecare sunt ireversibile. Deci un proces ireversibil în care există frecare este nestatic. Ne propunem să obţinem expresia analitică a celui de-al doilea principiu al termodinamicii în vederea aplicării sale ulterioare şi vom studia principiul al doilea al termodinamicii pentru procesele de echilibru (cvasistatice) şi procesele de neechilibru (nestatice).
4.4. Principiul al doilea al termodinamicii pentru procese reversibile
Rezultatele experimentale sintetizate în enunţul de bază al principiului al doilea şi în formulările echivalente au arătat ca nu se poate construi o maşină monotermă. Rămâne totuşi întrebarea: este posibilă conversia partială a Q în L şi în ce raport? Da, printr-o maşină termică a cărei substanţă de lucru, schimbând căldură cu doua sau mai multe termostate efectuează lucru mecanic. Spunem că un sistem termodinamic efectuează o transformare bitermă atunci când sistemul schimba caldură cu două termostate de temperaturi θ1şi θ2, R(θ1) şi R(θ2). Pentru transformări cicCarnot: raportul călduîntr-un proces ciclic bide natura sistemului, =temperatura empirică)
R(θ1)
L1
Q1
Q2
R(θ2)
Considerăm două sistefectuează simultan sacaldură cu acelaşi termotermostate. Repetând ciclul de n ocăldură fiind: nQ1, nQ2.Transformarea fiind reprimind atunci caldurile
lice reversibile biterme se poate demonstra prima teorema rilor schimbate de un sistem cu două termostate cu θ1 şi θ2 term reversibil, este acelaşi pentru orice sistem, nedepinzând fiind o funcţie universală de temperatura termostatelor (θ .
Fig.4.5. Schema maşinii biterme
Q1’
Q2’
L2
eme (corpuri de lucru) de natură diferită, M1şi M2 care u succesiv, procese ciclice biterme reversibile, schimbând stat ca în figura. Fie Q1 si Q2 căldurile primite de la cele doua
ri, obţinem o noua transformare ciclică bitermă, cantitatea de versibilă ciclul poate fi parcurs şi în sens invers, sistemele -Q1 şi -Q2. Repetând de asemenea, experienţa de n ori, există
38
pentru M1un ciclu reversibil pentru care căldurile au valorile -nQ1 şi -nQ2 cu n număr întreg mai mare sau mai mic decât zero. Considerăm apoi sistemul M2 care schimbă căldură cu aceleaşi două termostate, efectuând o transformare ciclică bitermă reversibilă. Dacă într-un ciclu schimbă căldurile şi Q atunci în urma repetării ciclului de m ori va schimba căldura m Q şi m , cu m de aceeaşi natură ca şi n.
Q'1Q2
'2'
'1Reunind cele doua sisteme M1şi M2 şi cele două cicluri biterme reversibile într-un singur ciclu biterm reversibil pentru sistemul M1+ M2, căldurile primite de la cele două termostate vor fi: nQ1+ m şi nQQ'1 2 + m Q , cu m si n numere întregi arbitrare mai mari sau mai mici decât zero.
2'
Putem alege n şi m astfel încât căldura schimbată de sistemul M1+ M2 cu cel de-al doilea termostat să fie zero:
nQ Q = 0 4.4 2'
2 + mstrict vorbind acest lucru nu este posibil decat dacă / QQ2
'2 este un număr raţional.
Raportul este un număr real, dar îl putem aproxima foarte bine printr-un număr raţional. Deci sistemul M1+ M2 efectuează o transformare monotermă ciclică reversibilă(!). Conform enunţului primar (ec. 4.1), înseamnă că şi căldura schimbată cu rezervorul R(θ1) = 0.
Ecuaţia 4.6 arată ca raportul căldurilor schimbate cu termostatele nu depinde de corpul (substanţa) de lucru. Deci:
QQ
f1
21 2= − ( ,θ θ )
)
4.7
f ( ,θ θ1 2
)
este o funcţie universală de temperatură, depinde numai de scara de temperatură aleasă iar semnul minus a fost ales ca sa fie pozitiv definită. Ecuaţia 4.7 constituie teorema Carnot. 4.4.2. Proprietăţile funcţiei f ( ,θ θ1 2
a) În 4.7 inversăm indicii: QQ
f2
12 1= − ( ,θ θ ) 4.8
Din 4.7 şi 4.8 se obţine:
ff
( , )( , )
θ θθ θ2 1
1 2
1= 4.9
b) f ( , )θ θ1 2 ≠ 0 Într-adevăr, dacă am avea pentru anumite valori ale lui θ θ1 2, , )f ( ,θ θ1 2 =0, atunci din 4.7 ar rezulta ca Q1 = -Q2 )f ( ,θ θ1 2 =0. Deci Q1 ar fi zero fără ca Q2 să se anuleze. Transformarea s-ar reduce la o transformare ciclică monotermă reversibilă, deoarece
39
schimbul de căldura cu R(θ1) este zero. Dar formularea de bază exclude această posibilitate.
)c) f ( ,θ θ1 2 ≠∝
1
4.11 proprietate care rezultă din a şi b. Presupunând-o continuă, rezultă că funcţia Carnot are mereu un semn constant, deoarece nu şi-ar schimba semnul decât trecând prin zero sau infinit. d) ),( =θθf Dacă θ θ1 , 2 sunt egale, transformarea este de fapt monotermă ciclică şi reversibilă şi căldura totală schimbată de cele două termostate de temperaturi egale este Q1+Q2. Conform cu formularea de bază a principiului al doilea, (Q1+Q2) = 0, de unde rezultă:
QQ
1
2
1= − 4.12
Comparând 4.12 cu 4.7 pentru θ θ θ1 2= = , rezultă: ),( θθf = 1 4.13
Din proprietatea (c) rezulta atunci ca funcţia Carnot nu poate lua decât valori mai mari ca zero. Din acest motiv s-a luat semnul minus în ecuaţia (4.7).
e) ),(),(
),02
012 θθ
( 1θθ
θθf ff
=
Consideră în afara termostatelor de temperatură θ θ1si 2 un al treilea de temperatură θ0 ca în figura 4.6.
Q2’
Q0’
Q2
Q1’
Q1
L2
L1 L3
Q0
R(θ2)
R(θ0)
R(θ1)
Fig.4.6. În acest caz teorema Carnot se scrie:
QQ
f
QQ
f
QQ
f
1
01 0
21 2
2
02 0
= −
= −
= −
( , )
( , )
( ,
''
'
'
θ θ
θ θ
θ θ )
4.14
În transformare, sistemul (M1+M2+M3) primeşte căldurile de la rezervoare după cum urmează: R( 1+ Q'1θ1) Q
R( 2+ 4.15 Q2'θ2) Q
40
R( ) Q Q0 0+ ' θ0
Se poate realiza masina termica astfel încât (Q1+ )=0, (QQ'1 2+ ) = 0 4.16 Q2
'
În acest caz dacă ecuaţiile 4.15 sunt satisfăcute, atunci transformarea este ciclică monotermă, reversibilă deoarece nu mai schimbă căldură decât cu R(θ
0
0). Conform cu formularea de bază a principiului al II-lea, rezultă:
Q Q0 + ' = 0, deci QQ
0
0
1'
= −
Ca urmare, ec.4.14 devin, din ec. 4.16:
),(
),(
),(
020
2
212
1
010
1
θθ
θθ
θθ
fQQ
fQQ
fQQ
−=
−=−
−=
4.17
Din ec.(4.17). împărţind membru cu membru prima şi ultima relaţie se obţine:
QQ
ff
1
2
1 0
2 0
=( , )( , )θ θθ θ
4.18
Comparând (4.18) cu cea de-a doua relaţie din 4.17 se obţine:
ff
f( , )( , )
( , )θ θθ θ
θ θ1 0
2 01 2= 4.18’
În care θ θ1si 2 sunt arbitrare. 4.4.3. Temperatura termodinamica absolută. Egalitatea lui Clausius
Fie o funcţie ϕ θ θ θ( ) ( , )= f 0 4.19 cu θ0 presupusă fixă şi θ lăsată să varieze. Din proprietatea (c) a funcţiei universale f ( , )θ θ 0 , rezultă caϕ θ( ) este mereu pozitivă, fără să se anuleze sau să devină infinită.
ϕ θ( ) >0 4.20 Relaţia 4.18’ se scrie:
ϕ θϕ θ
θ θ( )( )
( ,1
21 2= f ) 4.21
Relaţia (4.19) arata ca ϕ θ( ) depinde de alegerea lui θ0. Pentru altă alegere θ0’ avem o nouă funcţie:
′ = = =ϕ θ θ θθ θθ θ
ϕ θθ θ
( ) ( , )( , )( , )
( )( , )
'' 'f
ff f0
0
0 0 0 0
4.22
Deci noua funcţie nu diferă de vechea funcţie ϕ θ' ( ) ϕ θ( ) decât prin factorul constant
şi pozitiv 10 0f ( ,
( ,''
θ θθ θ
0 0
f)
= ) , conform proprietăţii (a). Reciproc, dacă înlocuim
funcţiaϕ θ( ) ϕ θ' ( )cu funcţia: = k ϕ θ( ) cu k>0, constantă, proprietăţile 4.20 şi 4.21 sunt valabile pentru . Funcţia ϕ θ' ( ) ϕ θ( ) se notează cu T şi se numeşte “temperatură
41
termodinamică” sau “temperatură absolută”, corespunzătoare temperaturii empirice θ, independentă de natura sistemului în studiu.
T = ϕ θ( ) 4.23 Marimea T este mereu pozitivă, (extremele T = 0 şi T →∝ sunt excluse) şi definită până la un factor constant pozitiv. Acest factor se fixează printr-o convenţie suplimentară. Această convenţie, acceptată internaţional, spune că punctul triplu al apei are temperatura absolută T = 273,16 K Se observă că relaţia 4.23 defineşte o scara de temperatură, dacă mai arătăm ca funcţia ϕ θ( ) este monoton crescătoare.(aceasta după ce vom demonstra inegaliatea Clausius) Ţinând seama de (4.21) şi (4.23), relaţia (4.7- teorema Carnot) devine:
QQ
TT
1
2
1
2
1
2
= − = −ϕ θϕ θ
( )( )
şi astfel: QT
QT
1
1
2
2
= −
deci: QT
QT
1
1
2
2
0+ = 4.24
cunoscută sub numele de “egalitatea lui Clausius”, adevărată pentru transformarea ciclică bitermă şi reversibilă. Ecuaţia 4.24 semnifică: suma algebrică a căldurilor reduse într-o transformare ciclică biterma reversibilă este zero.Ea este valabilă pentru orice ciclu Carnot reversibil. Ciclul Carnot Ciclul Carnot este descris de substanţa de lucru între două surse de căldură sursa caldă şi sursa rece. Corpul de lucru (de exemplu gazul) primeşte o cantitate de căldură de la sursa caldă a cărei temperatură Tsc este mai mare decât temperatura substanţei de lucru (Tsc>T1).Gazul se dilată producând lucru mecanic (de ex. deplasează un piston într-un cilindru).
4
3
1
Q2
V
p Q1
2
Această transmitere a căldurii către substanţa de lucru poate fi imaginată ca producându-se la o temperatură constantă a gazului (mai exact scăderea temperaturii gazului ca urmare a destinderii este compensată de aportul de căldură din exterior). Se realizează deci, o transformare izotermă la T1 = const. După ce gazul se destinde până la o anumită stare (2) aportul de căldură încetează aşa că destinderea gazului se face fără aport de căldură, deci adiabatic. În cursul destinderii adiabatice temperatura substanţei de lucru scade deoarece lucrul mecanic este produs pe seama scăderii energiei interne. După ce gazul a atins starea (3) de temperatură T2, destinderea care produce lucru mecanic încetează iar substanţa de lucru începe să revină la starea iniţială. Datorită
42
lucrului mecanic produs de la o sursă exterioară oarecare, gazul se comprimă şi în cursul acestei comprimări degajă o anumită cantitate de căldură care este transmisă sursei reci a cărei temperatură Tsr < T2. Acest transfer de căldură de la substanţa de lucru la sursa rece se produce astfel încât temperatura gazului în timpul comprimării rămâne constanta. Altfel spus, comprimarea se face urmând izoterma T2 = const. După ce gazul atinge starea (4) schimbul de căldură încetează dar gazul continuă să se comprime (adiabatic) până în starea iniţială. Deci ciclul Carnot se compune din două izoterme şi două adiabate. Lucrul mecanic produs de gaz este reprezentat de aria de sub curba 1-2-3, cel efectuat asupra gazului de aria de sub curbele 3-4-1 iar lucrul mecanic util furnizat unui receptor exterior este aria ciclului 1-2-3-4. Q1 este căldura furnizată substanţei de lucru de către sursa caldă, Q2 căldura cedată de substanţa de lucru sursei reci. Dat fiind că aportul de căldură de la sursa caldă la substanţa de lucru se produce în timpul transformării 1-2 la o diferenţă de temperatură constantă Tsc - T1 iar cedarea de căldură sursei reci în timpul transformării 3-4 tot la o diferenţă de temperatură finită T2- Tsr este evident ca transformările1-2 şi 3-4 sunt ireversibile.Ireversibilitatea tansformărilor poate fi redusă aprope la zero dacă diferenţa dintre temperatura corpului de lucru şi cea a sursei calde este infinit de mică: T1 = Tsc - dT T2 = Tsr + dT Dacă aceste condiţii sunt îndeplinite şi dacă destinderea gazului în 2-3 ca şi comprimarea sa în 4-1 se efectuează fără frecări, ciclul considerat devine reversibil. Într-adevăr să analizăm un ciclu Carnot descris de aceeaşi substanţă de lucru şi aceleaşi două surse însă în sens invers. Gazul se dilată urmând adiabata 1-4 şi efectuând un lucru mecanic. Temperatura scade în cursul destinderii adiabatice. După ce atinge starea (4) în care temperatura (TII) nu diferă decât cu o valoare mică faţă de cea a sursei reci, destinderea adiabatică încetează;
4
3
1
Q2
V
p Q1
2
TII = Tsr - dT Gazul se destinde, urmând izoterma 4-3 (TII = const) primind căldură de la sursa rece. În continuare asupra gazului se efectuează luceru mecanic din exterior în comprimarea adiabatică 3-2 şi gazul se încălzeşte.Starea (2) se alege astfel încât temperatura gazului (TI) să fie superioară temperaturii sursei calde cu o cantitate infinit mică: TI = Tsc + dT. gazul este comprimat în continuare, urmând izoterma TI ceea ce duce la starea sa iniţială (1). Compararea egalităţilor dintre temperaturi ne arată că T1=TI şi T2 = TII cu o precizie de până la o mărime infinit mică. Aceasta înseamnă că într-un ciclu Carnot invers starea substanţei de lucru se modifică într-un acelaşi interval de temperatură ca şi cea dintr-un ciclu Carnot direct. Lucrul mecanic pe ciclu va fi - L; semnul minus arată că lucrul mecanic este furnizat de către o sursă exterioară. Q2 este luată de la sursa rece şi transferată sursei calde. În plus, sursa caldă
43
mai primeşte şi o cantitate de căldură echivalentă lucrului mecanic pe ciclu furnizat din exterior : Q1 = Q2+ Lciclu. Deoarece T1=TI şi T2 = TII în ciclu invers sursa rece cedează ceeaşi căldură pe care o primeşte în ciclul direct iar sursa caldă primeşte aceeaşi Q1 pe care o furnizează în ciclul direct. Deci lucru mecanic consumat de o sursă exterioară pentru realizarea ciclului invers este acelaşi cu lucrul mecanic furnizat consumatorului exterior în ciclul direct. Aşadar, am realizat un ciclu Carnot invers, urmând exact drumul ciclului direct, adică într-un mod reversibil. Această reversibilitate a ciclului a fost obţinută datorită egalităţii (cu o precizie pâna la o mărime infinit mică) temperaturilor sursei calde şi substanţei de lucru şi sursei reci şi substanţei de lucru. Daca diferenţa de temperatură dintre sursa de căldură şi substanţa de lucru ar fi finită atunci ciclul ar fi ireversibil. Revenind la egalitatea lui Clausius, dacă ciclul ar consta din n transformări izoterme cărora le corespund sursele de căldura la temperaturile T1, T2,.......Tn,prin defalcarea în n-1 cicluri Carnot se poate generaliza relaţia (4.24). Orice ciclu reversibil care are o configuraţie oarecare poate fi reprezentat ca o sumă de cicluri Carnot elementare, fiecare fiind compus din două adiabate şi două izoterme.Primirea şi cedarea de căldură în fiecare dintre aceste cicluri elementare se realizează urmând izoterme. Aşa cum am mai spus, pentru aceasta este necesar să existe un număr infinit de mare de cicluri elementare. Suma ciclurilor Carnot elementare este perfect echivalentă cu un ciclu reversibil primar oarecare. Într-adevăr, deoarece adiabatele comprimării şi destinderii fiecărui ciclu Carnot elementar sunt dispuse la o distanţă infinit mică una de alta, fenomenele de schimb de căldură pot fi considerate izotermice; fiecare adiabată, cu excepţia celor două extreme, fiind parcursă de două ori şi în sensuri inverse, lucrul mecanic total al ciclului rămâne neschimbat, deoarece se afla pe cicluri Carnot elementare.
3 2
1
6 Fig. 4.7 QT
QT
QT
QT
1
1
4
3
2
2
5
3
0
0
+ =
+ = se adună şi rezultă:
QT
QT
Q QT
QT
QT
QT
1
1
2
2
4 5
3
1
1
2
2
3
3
0
0
+ ++
=
+ + =
Aşadar, se poate generaliza prin:
QT
i
ii
n
=∑ =
1
0 4.25
Numarul n de izoterme legate prin adiabate poate fi oricât de mare. În felul acesta pentru o transformare ciclică reversibila oarecare, ciclul se împarte într-o mulţime de cicluri Carnot infinit de mici (fig. 4.7) si suma (4.25) extinsa la întreg conturul ciclului, devine:
44
δQ
Trev∫ = 0 4.26
Aceasta este integrala lui Clausius pentru un ciclu reversibil. În această expresie T reprezintă temperatura sursei cu care vine în contact agentul termic (substanţa de lucru) pe o porţiune elementară a ciclului şi cu care schimbă caldura elementară δQ . Ciclul fiind presupus reversibil, temperatura T a sursei este egală cu temperatura agentului termic care evoluează în ciclu. 4.4.4. ENTROPIA. Forma generală a principiului al doilea pentru procese reversibile Ecuaţia (4.26) arată că mărimea δQ/T reprezintă o diferenţială totala exactă, aşadar, are proprietăţile unei mărimi de stare. Clausius i-a dat numele de entropie- S.
Deci, dS QT
=δ 4.27
Mărimea definită prin ecuaţia (4.27), numită entropie absolută are următoarele proprietăţi: • este mărime de stare aditivă, conservativă în procesele izentropice; • este definită pâna la o constantă arbitrară; • în cazul proceselor ciclice, variaţia entropiei este zero. Asemănarea dintre ecuaţia δQ= TdS şi δL = Ada permite interpretarea temperaturii ca o forţă generalizată termică a sistemului, S fiind un parametru de tip coordonată generalizată pentru procesul de transmisie a căldurii. Ecuaţia (4.27) constituie exprimarea cantitativă a principiului al doilea al termodinamicii, pentru procese cvasistatice reversibile: forma generală a principiului al II-lea pentru procese cvasistatice reversibile.
Ecuaţia δQT∫ = 0 constituie expresia matematică a principiului al II-lea al
termodinamicii pentru procese ciclice (aceasta exprimă univocitatea funcţiei S). Integrala lui dS de-a lungul unei curbe deschise (transformare reversibilă în care starea iniţială şi finală nu coincid) nu depinde decât de starea iniţială şi cea finală.
δ
σσ
σ
σ
σQT
dS S Srevf
i
f
i
f
∫ ∫= = −( ) (σ i ) 4.28
4.4.5. Consideraţii matematice în legătură cu caracterul olonom al căldurii Întrucât
λ ii
conform ecuaţiei calorice (de exemplu): căldura reprezintă o formă Pfaff de ordinul întâi se pune
problema cercetării caracterului ei olonom sau neolonom.
δQ C dT daA ai
i= + ∑
Fie o funcţie F(x,y), pentru care putem scrie: dyyxNdxyxMF ),(),( +=δ 4.29
cu M şi N funcţii continue derivabile. Teoremă: pentru ca expresia 4.29 să fie diferenţială totală exactă a unei funcţii de două variabile este suficient să fie satisfăcută relaţia:
xN
yM
∂∂
∂∂
= adică, ∂∂ ∂
∂∂ ∂
2 2Fy x
Fx y
= 4.30
45
Dacă xN
yM
∂∂∂
≠∂ atunci F este o formă Pfaff şi există întotdeauna o functie µ(x,y)
astfel încât produsul µ(x,y)δF admite o diferenţiala totală. µ(x,y) se numeşte factor integrant.
µ(x,y)δF = dG(x,y) = ∂∂
∂∂
Gx
dx Gy
dy+ 4.31
Ecuaţia µ(x,y)δF = 0 admite o familie de curbe integrale F*(x,y) = ct.; curbele se numesc adiabate. Formele Pfaff care admit diferenţială sau factor integrant se numesc olonome iar în caz contrar neolonome. Formele olonome conduc întotdeauna la o familie de adiabate (curbe sau suprafeţe) care este legată de o funcţie care admite diferenţială. Caldura elementară δQ reprezintă o formă Pfaff care pentru sistemele termodinamice simple este întotdeauna olonomă (admite factor integrant). Proprietatea de olonomie implica definirea entropiei: dS = µ(V,T)δQ; µ(V,T) =η(T)⋅ϕ(S), adică entropia este definită până la un factor arbitrar. η(T) = 1/T şi rezultă dS = δQ/T cu S = S(ai, T). 4.5. Principiul al II-lea al termodinamicii pentru procese ireversibile (nestatice) Dacă integrala δQ
T∫ = 0 (4.26) nu este nulă ci are o valoare, înseamnă că ciclul
este ireversibil. Cu cât valoarea este mai mare, cu atât ciclul este mai departe de reversibilitate. De aici rezultă ideea că variaţia de entropie poate fi folosită drept criteriu de apreciere a reversibilităţii. 4.5.1. Integrala lui Clausiu pentru procese termodinamice ciclice ireversibile Se consideră două maşini termice M1 şi M2 care lucrează după cicluri Carnot, folosind aceleaşi surse de căldură R(T1) şi R(T2).
R(T1)
Q1 Q1’
L1 M1 M2 L2
Q2 Q2’
R (T2)
Fig.4.8. M ă reversibilă 1 maşin M2 maşină ireversibilă Maşinile funcţionează astfel încât
agenţii termici respectivi primesc aceeaşi cantitate de căldură de la sursa caldă. În consecinţă, sistemul compus (M1 + M2) va parcurge de asemenea un proces ciclic biterm ireversibil, obţinut prin reunirea a n cicluri reversibile efectuate de M1 şi a m cicluri ireversibile efectuate de M2.
R(T1) R(T2) M1 nQ1 nQ2
46
M2 mQ1’ mQ2’ M1 + M2 nQ1+ mQ1’ nQ2 + mQ2’ Punem condiţia nQ2 + mQ2’= 0; atunci transformarea sistemului M1 + M2 este monotermă. Dat fiind că transfomarea lui M2 este ireversibilă, atunci întreaga transformare a sistemului M1 + M2 este ireversibilă şi avem conform cu formularea primară a principiului al II-lea:
pentru că T1≠ 0 , înlocuind 4.33 în 4.32 se obţine:
mQTT
mQ11
22 0' '+ .34 < 4
De unde se poate scrie: QT
QT
1
1
2
2
0' '
+ < 4.35
Relaţia 4.35 poate fi extinsă la orice ciclu format dintr-o mulţime de cicluri Carnot ireversibile:
QT
i
ii
n
=∑ <
1
0 4.36
Fig.4.9 Ciclu oarecare ireversibil divizat în cicluri Carnot elementare ireversibile
p
δQi,1 b a M c
i,2 d
V
N δQ
În cursul proceselor elementare ab şi cd, temperatura variază infinitezimal cu dT. Dacă neglijăm variaţia infinitezimală dT, atunci ab şi cd sunt izoterme ireversibile, pentru că aparţin ciclului ireversibil. Transformările ad şi cb sunt prin ipoteză, transformări adiabatice reversibile. Deci pentru ciclul Carnot elementar ireversibil abcd, funcţia Carnot se scrie:
δ δQ
TQT
i i, ,1
1
2
2
0+
< 4.37
Pentru toate ciclurile infinitezimale Carnot care alcătuiesc ciclul ireversibil oarecare se obţine:
δQT
i
ii
n
=∑ <
1
0 4.38
Procesul de trecere la limită (n→∝) va conduce în cazul transformării ciclice ireversibile la integrala:
47
δQ
Tirev∫ < 0 4.39
Aceasta este integrala Clausius pentru un ciclu ireversibil oarecare. Ea este negativă spre deosebire de cazul proceselor ciclice reversibile când este zero.
4.5.2. Forma generală a principiului al II-lea al termodinamicii pentru procese ireversibile
Ecuaţia (4.39) este importantă pentru că ea conduce la formularea matematică a principiului al doilea pentru procese ireversibile, oferind un criteriu cantitativ de apreciere a ireversibilităţii. Să considerăm o transformare ireversibilă între stările σ1→σ2 (Fig.4.10) şi o transformare reversibilă σ2→σ1.
δ δ δ
σ
σ
σ
σQT
QT
QT
irev irev rev∫ ∫ ∫= +2
2
1
0 4.40 <1
1σ
2σ
p Fig.4.10: σ2→σ1transformare reversibilă σ1→σ2 transformare ireversibilă V Dar pentru procesul reversibil σ2→σ1, rezultă:
δ δ
σ
σ
σ
σQT
QT
rev rev
2
1
1
2
∫ ∫= − 4.41
Înlocuind (4.41) în (4.40) se obţine:
δ δ
σ
σ
σ
σQT
QT
irev rev
1
2
1
2
0∫ ∫− <
adică:
δ δ
σ
σ
σ
σQT
QT
irev rev
1
2
1
2
∫ ∫< 4.42
Dar conform cu cele arătate anterior, δ
σ
σ QT
rev
1
2
∫ reprezintă variaţia mărimii de stare -
entropia.
Astfel: )( 12
2
1
SST
Qirev −<∫σ
σ
δ 4.43
Aceasta este formularea matematică a principiului al II-lea pentru procese ireversibile. Ea afirmă: în transformările ireversibile valoarea Integralei lui Clausius este mai mică decât variaţia entropiei. Principiul al II-lea poate fi exprimat, în general, astfel:
48
( )S S QT2 1
1
2
− ≥ ∫δ 4.44
sau pentru un proces elementar:
dS QT
≥δ 4.45
Aceasta arată că entropia poate constitui o măsură a gradului de ireversibilitate a proceselor termodinamice. În cazul sistemelor izolate adiabatic, 4.45 devine:
dS ≥ 0 , 4.46 S S2 ≥ 1
≥
În cazul proceselor adiabatice, în general pentru sisteme izolate adiabatic, în care se desfăşoară procese reversibile sau ireversibile, entropia rămâne constantă sau nu poate decât să crească. Adică pentru un sistem izolat, integrala Clausius este nulă, deoarece sistemul nu schimbă căldură cu mediul ambiant şi deci:
( ) .S S sist izolat2 1 0− 4.46’ • Entropia unui sistem izolat nu poate să scadă; ea se menţine constantă dacă în sistem se desfăşoară numai procese reversibile şi creşte dacă în sistem au loc procese ireversibile.
Dacă în starea iniţială sistemul se află în echilibru termodinamic intern, entropia va rămâne constantă în timp. În cazul în care, starea iniţială a sistemului termodinamic este de neechilibru termodinamic, în sistem se desfăşoară procese spontane ireversibile, care tind să aducă sistemul într-o stare de echilibru termodinamic; în acest caz, conform cu ecuaţia 4.46’, entropia va creşte, tinzând către o valoare finală maximă. Odată atinsă această valoare, sistemul va rămâne în echilibru pâna la eventuala ridicare a izolării, ceea ce se exprimă prin:
dS = 0 d S2 0< 4.47 Într-un sistem izolat, echilibrul presupune egalizarea temperaturilor tuturor
corpurilor care alcătuiesc sistemul; după stabilirea echilibrului nu se mai poate produce în sistem transformarea căldurii în lucru mecanic, deoarece lipsesc sursele de căldură de temperaturi diferite. Deci creşterea entropiei unui sistem izolat reprezintă o măsură a degradării energiei, adică a reducerii capacităţii de producere a lucrului mecanic în interiorul sistemului.
4.5.3. Sursa de entropie Cu ajutorul entropiei ca funcţie de stare, definită pe baza principiului al doilea,
se poate stabili sensul în care se desfăşoară procesele în natură. În cazul sistemelor neizolate, entropia variază atât datorită ireversibilităţii proceselor cât şi datorită schimbului de căldură cu exteriorul. Astfel, dacă exprimăm principiul al II-lea sub forma:
TdS Q− ≥δ 0 4.48 unde δQ este căldura schimbată de sistem cu mediul exterior; notăm cu δ δQ TdS Q= − , căldura necompensată cum a fost numită de Clausius şi ecuaţia (4.48), devine (δQ intervine în procesele ireversibile şi este zero în cele reversibile):
49
δQ ≥ 0 4.49 ţinând seama de această notaţie, variaţia de entropie va fi:
dS QT
QT
d S d Se i= + = +δ δ 4.50
unde deS se datoreşte schimbului de căldură δQ cu mediul exterior iar diS reprezintă entropia produsă datorită creerii de căldură necompensată δQ în interiorul sistemului în urma unui proces care se desfăşoară ireversibil. Deci, din (4.50) rezultă că entropia unui sistem variază fie datorită transportului de entropie de la sistem spre mediu sau din mediu spre sistem (deS), fie prin crearea ei în sistem (diS). Variaţia entropiei externe, deS poate să fie pozitivă, negativă sau nulă, în funcţie de interacţia sistemului cu mediul. Astfel, pentru un sistem izolat adiabatic, care nu schimbă nici materie nici căldură cu mediul exterior, deS=0 iar diS≥0 !! Entropia poate fi creată în sistem dar nu poate fi distrusă. În cazul sistemelor deschise care pot schimba atât caldură cât şi materie cu mediul, semnul entropiei externe este identic cu semnul cantităţii de căldură schimbată cu mediul exterior, în acord cu convenţia generală de semn pentru pierdere sau câştig de energie. Entropia creată în sistem este întotdeauna pozitivă; în procese ireversibile se creează entropie, nu se distruge (diS>0). Dacă sistemele sunt izolate: dS= diS
diS≥0 4.51 În cazul proceselor cvasistatice diS=0, iar variaţia entropiei sistemului este egală cu entropia externă. O problemă fundamentală în termodinamica proceselor ireversibile este exprimarea variaţiei de entropie în timp:
dSdt
d Sdt
d Sdt
e i= + 4.52
Primul termen al membrului al doilea reprezintă viteza schimbului de entropie cu mediul, iar al doilea termen reprezintă viteza cu care se produce entropie în sistem, în urma unui proces. Se defineşte în felul acesta, “sursa de entropie” (producţia de entropie), P, ca entropia produsă în unitatea de timp şi volum, într-o zonă dată a unui sistem:
Pd Sdti= 4.53
O transformare ireversibilă este caracterizată de o valoare pozitivă a sursei de entropie. Valoarea sa scade în cursul unui proces. Ecuaţia de bilanţ pentru sursa de entropie este:
P≥0 4.54 Într-o transformare ireversibilă sursa de entropie este totdeauna şi pretutindeni mai mare decât zero, iar într-un proces reversibil ea se anulează.
4.5.4. Formulări echivalente ale principiului al II-lea al termodinamicii Sintetizând consideraţiile privind transformarea monotermă şi bitermă susţinute de rezultate experimentale certe, se pot da mai multe enunţuri ale principiului al II-lea al termodinamicii care sunt echivalente sau se completează reciproc, după cum urmează:
50
• Este imposibil să se construiască o maşină monotermă sau un perpetuum mobile de speţa aII-a.
• Este imposibil un proces ciclic în care căldura să treacă de la un termostat cu temperatură scăzută la un termostat cu o temperatură ridicată, fără nici-o modificare în mediul exterior (Clausius).
• Nu este posibil un proces ciclic al cărui unic rezultat să fie efectuarea de lucru mecanic pe seama scăderii energiei interne a unui singur termostat (Thomson).
• În vecinătatea oricărei stări de echilibru a unui sistem termodinamic, omogen, există stări care nu pot fi atinse în mod adiabatic (principiul inaccesibilităţii adiabatice - Caratheodory). Acest enunţ reprezintă faptul că adiabatele nu se intersectează, aşa cum principiul zero afirma că izotermele nu se intersectează.
• Orice sistem termodinamic este caracterizat de o funcţie univocă de stare numită
entropie, care într-un proces elementar satisface relaţia dS QT
≥δ , conţinând
afirmaţia: pentru sistemele izolate adiabatic în care se desfăşoară procese reversibile sau ireversibile, entropia rămâne constantă sau nu poate decât să crească.
Analizând conţinutul principiilor I şi II se poate realiza o comparaţie între U şi S: a) Energia internă U se conservă pentru sisteme izolate; cu ajutorul ei nu se poate face distincţie între stările iniţială şi finală, adică nu poate fi indicat sensul în care se desfăşoară procesle. S, dimpotrivă, pentru sisteme izolate adiabatic în care se desfăşoară procese ireversibile creşte indicând sensul lor de desfăşurare. b)U nu poate fi creata şi nici distrusă. S poate fi creată în procesle ireversibile dar nu poate fi distrusă. c) Creşterea entropiei în procese ireversibile indică faptul ca energia internă se depreciază, devine din ce în ce mai puţin accesibilă transformării în lucru mecanic. L maxim se obţine în procese reversibile (ideale) în care S rămâne constantă. d) Principiul I este legat de posibilitatea transformării integrale a L în Q, stabilind echivalenţa cantitativă a celor două mărimi, iar principiul al II-lea este în legătură cu posibilitatea transformării parţiale a Q în L, afirmând neechivalenţa calitativă a acestor mărimi. e) Principiul al II-lea este de natură statistică având sens numai pentru sisteme macroscopice finite în spaţiu şi timp şi nu are acelaşi grad de generalitate ca principiul I. Completare la temperatura termodinamică absolută Acum suntem în măsură să demonstrăm ca T este o funcţie crescătoare de temperatura empirică. Fie deci doua surse termice de temperaturi θ1 şi θ2 şi funcţiile: T1=ϕ(θ1) şi T2==ϕ(θ2). Presupunem că θ1 > θ2 Fie A un sistem care nu poate interacţiona cu mediul decât termic şi în starea iniţială are temperatura θ2. Dacă punem sistemul A în contact cu un termostat de temperatură θ1, atunci A primeşte o căldură Q1 până îşi atinge noua stare de echilibru (Q1>0). Apoi, dacă readucem sistemul A în contact cu θ2, atunci A va ceda termostatului de temperatură θ2 căldura Q2<0 şi revine în starea sa iniţială. Deci ∆U = 0. Cum deja am presupus că L=0, se obţine: Q1+Q2=0 şi Q2=-Q1.
51
Procesul fiind biterm şi ireversibil, se poate scrie inegalitatea lui Clausius:
QT
QT
1
1
2
2
0+ <
de unde : QT T1 1 01 2
−
< şi T1> T2 deoarece Q > 0
Deci: θ1 > θ2 determina T1> T2 tocmai ceea ce trebuia demonstrat. Monotonia temperaturii termodinamice ne confirmă ca relaţia de definiţie T = ϕ θ( ) defineşte o scară de temperatură.
52
5. APLICATII ALE PRINCIPIILOR TERMODINAMICII
5.1.Randamentul motoarelor termice După cum am văzut, al doilea principiu al termodinamicii a fost stabilit din analiza funcţionării maşinilor termice.Ţinând seama că schimbul de caldură dintre corpul de lucru şi termostate este reverşibil numai dacă are loc în mod izoterm, şi că ciclul Carnot este o transformare reprezentată prin două izoterme şi două adiabate, aşa cum am conşiderat anterior, putem reprezenta ciclul astfel:
T 1 T1 Q1 2
T2 4 Q2 3 S
Subliniem faptul că dacă un sistem efectuează o transformare ciclică,
schimbând căldură cu două termostate de temperaturi date, atunci unicul ciclu reverşibil este ciclul Carnot. Maşina bitermă al cărui corp de lucru efectuează un ciclu reverşibil se numeşte maşină Carnot, iar toate celelalte sunt maşini biterme ireverşibile. Presupunem că T1> T2, adică R(T1) - sursa caldă (încălzitor) şi R(T2) - sursa rece (răcitor); Q Q1 > .2 Q1 este primită de la R(T1) şi Q2 este cedată sursei rece, lucrul mecanic efectuat pe fiecare ciclu fiind: L = Q1- Q2 Pentru a caracteriza eficienţa funcţionării unei astfel de maşini se introduce noţiunea de randament termic, care se defineşte ca:
η = =lucrulmecanicpeciclucalduraprimitapeciclu
LQ1
Conform principiului al doilea δQT∫ = 0 adică δ δ δ δQ
TQT
QT
QT1
2
2
3
3
4
4
1
0∫ ∫ ∫ ∫+ + + =
Integralele 2-3 şi 4-1 sunt zero pentru că transformările sunt adiabatice şi rezultă:
.02
2
1
1 =−TQ
TQ
Ca urmare, η =−
= −Q Q
QTT
1 2
1
2
1
1 5.1
• Din expreşia randamentului se observă că acesta nu depinde de substanţa de lucru ci este determinat numai de temperaturile rezervoarelor termice (prima teoremă Carnot).
• Randamentul ciclului Carnot mai mic decât 1 arată că în natură nu se poate realiza decât converşia parţială a căldurii în lucru mecanic şi că acest proces implică în mod
1
obligatoriu cedarea unei părţi Q2 din căldura primită de substanţa de lucru, la o temperatura T2< T1. Converşia parţială a căldurii în lucru mecanic arată la rândul său că acţiunea termică este mult deosebită de alte tipuri de acţiuni care se manifestă în natură. În urma proceselor ciclice, orice tip de acţiune se poate transforma integral în acţiune termică, dar acţiunea termică se transformă doar parţial în alte tipuri de acţiuni, conform principiului al II-lea. De aici, afirmaţia că principiul al doilea arată neechivalenţa calitativă dintre căldură şi lucru mecanic. • Dacă maşina bitermă funcţionează după un ciclu oarecare ireverşibil, atunci,
ţinând seama de inegalitatea lui Clausius relaţia 1 conduce la : 12
1
2
1
− < −QQ
TT
η iTT
< −1 2
1
5.2
Deci, randamentul maşinii termice ireverşibile care funcţionează, schimbând căldură cu aceleaşi termostate ca maşina Carnot, este întotdeauna mai mic decât randamentul maşinii Carnot. Expreşia 5.2 reprezintă conţinutul celei de-a doua teoreme Carnot: randamentul oricărei maşini biterme care funcţionează cu aceleaşi termostate de temperaturi T1 şi T2, nu poate depăşi randamentul maşinii ideale Carnot. Cu alte cuvinte, ireverşibilitatea diminuează randamentul de converşie a căldurii în lucru mecanic, contribuind în plus la “degradarea” energiei interne. Pentru refacerea calităţii energiei interne este necesară totdeauna o compensaţie. • Randamentul maşinilor termice în afară de faptul că nu poate avea valoarea 1, are
o limită superioară impusă de capacitatea energiei de a se transforma dintr-o formă într-alta, de neechivalenţa calitativă dintre căldură şi lucru mecanic.
• Din relaţia (5.2) rezultă că randamentul maşinii Carnot este subunitar şi cu atât mai mare cu cât T2/T1 este mai mic.
Se poate calcula care din cele două surse influenţeaza mai mult randamentul: ∂η∂T
TT1
2
12= ∂η
∂T TTT2 1
1
12
1= − = − şi întrucât T1>T2 rezultă ∂η
∂∂η∂T T1 2
<
Deci, variatia temperaturii sursei calde influenteaza mai puţin randamentul ciclului Carnot prin comparaţie cu variaţia temperaturii sursei reci. Altfel spus, randamentul creşte dacă scade temperatura sursei reci. ηciclului Carnot este egal cu 1 numai dacă Q2=0 şi T2=0, în contradicţie cu formularea de bază a principiului al II-lea al termodinamicii. În cazul în care Q2=0 şi T2=0, izoterma se confundă cu adiabata şi ciclul nu se poate închide. Cu alte cuvinte nu este poşibil un ciclu Carnot cu rezervorul rece în stare de zero absolut. Dupa destinaţia lor, maşinile termice se împart în trei tipuri principale: -motoare termice -pompe termice (pompe de caldură) -maşini frigorifice Motorele termice, despre care am vorbit, transformă Q în L; pompele termice pe seama unui L cheltuit şi a căldurii luate de la mediu (cu temperatura mică), încălzesc corpuri cu temperatura mai mare; maşini frigorifice care cu ajutorul lucrului mecanic, L cheltuit iau caldura Q de la corpul care se răceşte şi o transmite mediului înconjurător. Maşina Carnot poate funcţiona şi ca maşină frigorifică.
2
Corpul de lucru pe seama lucrului mecanic efectuat de mediul exterior, primeşte căldura Q2 de la sursa rece şi transmite într-un ciclu căldura Q1=Q2+L, sursei calde.
T 1 Q1 2 T
T2 4 Q2 3 S
1
Eficienţa unei maşini frigorifice este determinată de coeficientul de răcire:
ψ = =−
=−
QL
QQ Q
TT T
2 2
1 2
2
1 2
5.3
şi poate fi o mărime subunitară, supraunitară sau zero. Eficienţa pompei de căldură:
ϕ = =−
QL
TT T
1 1
1 2
cu T1 > T2 5.4
5.2. Relaţia fundamentală a termodinamicii. Aplicaţii Principiul zero introduce temperatura T ca parametru de stare şi pentru
sistemele la echilibru arată că forţele generalizate sunt funcţii de coordonatele generalizate şi temperatură şi la fel energia internă:
( )TaaaAA nii ,,......, 21= şi ( )TaaaU n ,,......, 21=U Principiul I : exprimarea matematică infinitezimală: dU = δQ - δL şi finită:
cu convenţia că: pentru Q>0 sistemul primeşte căldură;Q<0, sistemul cedează căldură;L>0, sistemul efectuează lucrul mecanic; L<0, asupra sistemului se efectueaza lucrul mecanic.
LQU −=∆
Principiul al II-lea: exprimarea matematică infinitezimală dS ≥ δQT
Combinând expreşiile matematice ale principiului I şi al II-lea se obţine:
dS dU LT
sau dU TdS L≥+
≤ −δ δ, , 5.5
Deci ecuaţia dSdU A da
T
i ii≥
+ ∑ 5.6
reprezintă ecuatia fundamentala a termodinamicii; egalitatea este valabila în cazul proceselor termodinamice reverşibile iar inegalitatea în cazul celor ireverşibile.Comparând expreşiile, pentru procese cvaşistatice: TdS= δQ cu δL =
3
A dai ii
∑ , rezultă că dacă într-o diagramă (p,V) , δL = pdV reprezinta aria de sub
curba de transformare, în coordonate (T,S) aria de sub curba va reprezenta căldura.
Avantajele utilizarii diagramei (T,S) • este comodă în sensul că ea reprezintă imediat şi sugestiv atât caldura schimbată în cursul ciclului cât şi lucrul mecanic produs de corpul de lucru într-un ciclu:
ariaQQLciclu =−= 21 permite să se vadă în ce porţiuni ale ciclului caldura este furnizata substanţei de
lucru şi în ce porţiuni ea este primită de la substanţa de lucru: unui aport reverşibil de căldură îi corespunde o creştere de entropie, iar unei cedări de căldură, o micşorare a entropiei.
•
5.2.1. Relaţii diferenţiale de dependenţă între funcţiile de stare şi parametri de stare pentru un sistem termodinamic şimplu
Utilizând relaţia fundamentală a termodinamicii pot fi stabilite relaţii diferenţiale de dependenţă între funcţiile de stare pentru un sistem termodinamic şimplu: 10 (V,T) : U = U(V,T) p = p(V,T)
S = S(V,T) dU = TdS – pdV
⇒
∂∂
+
∂∂
=
∂∂
+
∂∂
=
VVST
TSS
VVUT
TUU
TV
TV
ddd
ddd⇒
∂∂
+
∂∂
=
+
∂∂
+
∂∂
=
VVST
TSS
VVUT
TU
TS
TV
TV
ddd
dpdd 1
+
∂∂
=
∂∂
∂∂
=⇒
∂∂
=
∂∂
pVU
TVS
TSTC
TU
TTS
TT
VV
VV
1
1ν
5.7.
4
Deoarece S(V,T) şi U(V,T) sunt diferenţiale totale exacte, TVS
VTS
∂∂∂
=∂∂
∂ 22
şi
TVU
VTU
∂∂∂
=∂∂
∂ 22
;
Foloşind aceste identităţi se obţine:
pTpT
VU
VT
−
∂∂
=
∂∂ 5.8.
vT T
PVS
∂∂
=
∂∂ care reprezintă o relaţie Maxwell. 5.9.
20 (p,T) : H = H(p,T)
V = V(p,T) S = S(p,T) dU = TdS – pdV
⇒
∂∂
+
∂∂
=
∂∂
+
∂∂
=
ppST
TSS
ppHT
THH
Tp
Tp
ddd
ddd
⇒
∂∂
+
∂∂
=
−
∂∂
+
∂∂
=
ppST
TSS
ppHT
TH
TS
Tp
Tp
ddd
dVdd 1
−
∂∂
=
∂∂
∂∂
=⇒
∂∂
=
∂∂
VpH
TpS
TSTC
TH
TTS
TT
pp
pp
1
1ν
5.10
Deoarece S(p,T) şi H(p,T) sunt diferenţiale totale exacte, TpS
pTS
∂∂∂
=∂∂
∂ 22
şi
TpH
pTH
∂∂∂
=∂∂
∂ 22
;
Foloşind aceste identităţi se obţine:
pT TVTV
pH
∂∂
−=
∂∂ 5.11
pT TV
pS
∂∂
−=
∂∂ a doua relaţie Maxwell. 5.12
5.2.2. Calculul energiei interne, entalpiei şi entropiei • Energia
( ) dVdTνC dU TV, U U V
−
∂∂
+== pTpT
V
5
( ) ∫∫
−
∂∂
+= dVdTCνTV, U V pTpT
V 5.13
Ca să se rezolve ecuaţia integralo-diferenţială trebuie cunoscută dependenţa lui CV de temperatură şi ecuaţiile de stare ale sistemului termodinamic.
Pentru gazul ideal: CV = ct, VνR
=
∂∂
VTp
rezultă 0=
∂∂
TVU
U(T) = νCVT + U0
•
5.14
Entalpia H = H(p,T)
dpTVTVdTdp
pHdT
THdH
pTp
∂∂
−+=
∂∂
+
∂∂
= pνC
( ) dpTVTVdTTpH
p∫∫
∂∂
−+= pCν, 5.15
Pentru gazul ideal: Cp = ct, p
νR=
∂∂
pTV
rezultă 0=
∂∂
TpH
Ca urmare, H(p,T) = νCpT+H0
•
Entropia ),( TVS
dV;dTT
νC dS deci dV dT dS
V
V
TV
∂∂
+=
∂∂
+
∂∂
=Tp
VS
TS
rezultă dVTp
TdTS
V∫
∂∂
+∫=∆ VCν 5.16
),( TpS
;dpdTT
νC dS deci dp dT dS
p
p
Tp∫
∂∂
−=
∂∂
+
∂∂
=TV
pS
TS
rezultă dpTV
TdTS
p∫
∂∂
−∫=∆ pCν 5.17
)pS ,( V
;- dS deci dp dV dS VpVp
dVTpdp
TV
pS
VS
∂∂
+
∂∂
=
∂∂
+
∂∂
=
rezultă ( ) dpTVdV
TppVS
p∫
∂∂
−∫
∂∂
=∆V
, 5.18
Pentru gazul ideal, expreşiile variaţiei de entropie se pot calcula, ţinând seama de ecuaţia de stare RTpV ν= şi de faptul că CV=const şi Cp=const.
∫ ∫+=∫
∂∂
+∫=∆VdVRTdCdV
Tp
TdTS V
V
lnCν V ν
0lnln SVRTCS V ++= νν 5.19
6
∫ ∫−+∫
∂∂
−∫=∆p
dpRTdCdpTV
TdTS p
p
lnCν p ν
0lnln SpRTCS p +−= νν 5.20
( ) ∫−∫=∫
∂∂
−∫
∂∂
=∆p
dpRVdVRdp
TVdV
TppVS
p
ννV
,
0lnln SpRVRS +−= νν 5.21
5.3.Procese termodinamice fundamentale Procesele în care o marime termodinamica îşi pastreaza valoarea constantă sunt conşiderate procese termodinamice fundamentale datorită importanţei lor teoretice şi aplicative. Astfel de procese sunt: procese adiabatice, caracterizate prin Q=0; procese politropice, caracterizate prin capacitatea calorica constanta; procese izoterme, caracterizate prin temperatura constanta; procese izocore, caracterizate prin volum constant; procese izobare, caracterizate prin preşiune constantă. Vom studia principalele transformări termodinamice în vederea stabilirii legilor care le guvernează pentru sisteme termodinamice şimple.
5.3.1.Procese adiabatice Se numeşte transformare adiabatică transformarea termodinamică în cursul căreia sistemul nu primeşte şi nici nu cedează căldură: δQ = 0. În condiţii reale o transformare este adiabatica daca sistemul este “înzestrat” cu o bună izolaţie termică sau dacă destinderea (sau comprimarea) gazului se face atât de rapid încât, practic nu are loc nici un fel de schimb de căldură între sistem şi mediu. Deoarece pentru o transformare reverşibilă principiul al doilea TdS = δQ, în transformarea adiabatica dS = 0. Cu alte cuvinte o transformare adiabatică reverşibilă este în acelaşi timp şi izentropică. Transformarea adiabatică poate fi şi ireverşibilă.De exemplu curgerea unui gaz real printr-un tub rugos, înzestrat cu înveliş adiabatic care nu permite schimbul de caldură. Curgerea gazului va fi, în consecinţă, adiabatică pentru că nu primeşte şi nici nu cedează căldură. Curgerea unui gaz real într-un tub rugos fiind însoţită întotdeauna de frecare, care produce o dişipare de energie de către fluidul care curge, această transformare este ireverşibilă şi ca orice proces ireverşibil antrenează o creştere a entropiei: TdS > δQ. În cazul transformarii adiabatice ireverşibile examinată, δQ = 0, dar dS>0, deci transformarea adiabatică nu este şi izentropică. Prin urmare se poate spune că orice transformare izentropică a unui sistem izolat este adiabatică, însă reciproca nu este adevarată decât în cazul transformarilor reverşibile. În acest paragraf ne referim la transformarile reverşibile (dS =0) şi vom analiza relaţiile dintre parametrii diverselor stări într-o transformare adiabatică reverşibilă. Pentru aceasta vom deduce ecuaţia diferenţială a unei transformari izentropice. δδQ dU pdVQ dH Vdp
= += −
Daca transformarea este izentropică,
7
VdpdHpdVdU
=−=
de unde
VpHp
VU
SS
=
∂∂
−=
∂∂ ;
Vp
pHVU
S
S −=
∂∂
∂∂
5.22
Cu ajutorul acestor ecuaţii nu este greu să se obţină relaţia:
SS V
ppV
UH
∂∂
−=
∂∂ 5.23
Aceasta relaţie nu este decât ecuaţia diferenţialăa unei transformări izentropice. Ea exprimă variaţia mărimilor calorice H şi U ale sistemului în funcţie de mărimile sale termice p şi V într-o transformare izentropică.
Se noteaza cu k marimea : SU
Hk
∂∂
= 5.24
Marimea k se numeste exponentul transformarii izentropice, sau exponent izentropic. Ţinând cont de aceasta notaţie, relaţia ( 5.23) devine:
kdlnV- dlnp ;lnln-sau ==
∂∂
=
∂∂
− kVpk
Vp
pV
SS
5.25
Această ecuaţie diferenţială stabileşte o relaţie între p şi V într-o transformare izentropică. Integrând această relatie între două stări ale transformarii izentropice se obţine :
2
1
1
2
1
2
1
2 lnlnsau lnlnVVk
pp
VVk
pp
=−= 5.26
Dacă în intervalul conşiderat exponentul izentropic ramâne constant, el poate fi scos de sub integrală şi astfel obţinem:
kkk
k
VpVpVV
pp
22111
2
1
2 lnln =⇒−= 5.27
Aşadar, pentru fiecare stare a sistemului care parcurge o transformare izentropica (cu k constant) avem:
PVk = const 5.28 Aceasta ecuaţie este ecuaţia adiabatei, a lui Poisson. Dacă exponentul adiabatic variază când sistemul îşi modifică starea şi dacă se cunoaşte variaţia lui, atunci integrala se rezolvă numeric pentru a găşi valoarea lui p din valorile parametrilor din starea iniţială şi volumul din starea finală. Se mai poate foloşi şi valoarea medie a lui k şi atunci ecuaţia Poisson este :
unde .constpV mediuk =
1
2ln
ln2
1
VV
Vkdk
V
Vmediu
∫= 5.29
Subliniem ca ecuatiile transformarii izentropice sunt aplicabile atât pentru gaze cât şi pentru lichide şi solide (n-am facut nici-o presupunere privind şist termodinamic).
8
Exponentul izentropic capata o valoare senşibil diferita pentru diversele faze de stare ale substantei. Astfel, pentru lichide şi solide k este foarte mare şi sufera o variatie substantiala o data cu temperatura. Pentru apa la 00C k=3 602 000 în timp ce la t=500C, k=187 000 iar la t=1000C k=23000. Pentru gaze şi vapori k variaza (se micsoreaza) putin cu temperatura; pentru majoritatea gazelor el este cuprins între 1,3 şi 1,7; k variaza puternic numai în vecinatatea curbei limita. În Fig... sunt reprezentate valorile exponentului izentropic k pentru vaporii de apa.
Pentru exponentul adiabatic al gazelor se deduce expreşia ca fiind raportul dintre căldurile specifice sau molare la preşiune constantă şi volum constant:
Din TV
p
SSv
p
s
T
S Vp
CC
pVk
VkVp
CC
kk
Vp
pVk
∂∂
⋅−=⇒−=
∂∂
=
∂∂
−= 1 ; relatiadin si
Vp
Vp
Vp
CC
Vp
TTV
p
S
−=
∂∂
∂∂
=
∂∂ dar
şi ca urmare γγ =⇒= perfectk V
p
CC
5.30
Caldurile specifice şi molare ale gazului perfect variază foarte puţin în timp;de aceea, valoarea lui γ poate fi conşiderată cu buna precizie, practice independenta de temperatură. În ceea ce priveste relaţia dintre temperatura a doua stări într-o transformare izentropică se poate scrie , ţinând seamai de ecuaţia Poisson, pVk =
const.,TV sau constk =−1 .1
constkk
=−
Tp Prin urmare, în cursul unei destinderi izentropice (adiabatic reverşibilă) sistemul se răceşte,deci destinderea adiabatică reverşibilă este un procedeu eficient de răcire a gazelor ( !). • Lucrul mecanic în destinderea adiabatică δQ = dU + pdV; pentru transformarea izentropică pdV = -dU valabilă atât pentru transformarea reverşibilă
cât şi ireverşibilă.
( 12
2
121 ; UULpdVL 12 −−== ∫− )
Ecuaţiile de mai jos nu pot fi foloşite decât în cazul când exponentul izentropic este constant sau se ia o valoare medie a acestuia.
−
−=
−
−==⇒=
−−
∫k
kk
k
k
k
k
pp
kVpL
VV
kVpdV
VVpL
VVpp
1
1
21112
2
1
1
2
1111112
11 1
1sau 1
15.31
9
Dat fiind că pentru un gaz perfect energia internă nu depinde decât de temperatură şi caldura specifica este constantă se obţine expreşia pentru lucru mecanic:
( ) ( 22111212V12 11sau νCν 2
1
VpVpLTTdTCLT
T V −−
=−== ∫ γ) 5.32
Variaţia de entropie este nulă în procesele adiabatice reverşibile, şi căldura molară de asemenea.
5.3.2.Transformari politropice Se numesc transformari politropice transformările pentru care C este constant şi aceste transformări satisfac ecuaţia:
.constpV n = 5.33 pentru o valoare n arbitrară, constantă pentru transformarea conşiderată. n poartă numele de coeficient politropic şi poate lua orice valori între -∝ şi ∝ . Noţiunea de transformare politropică a fost introdusă prin analogie cu transformarea adiabatică ; diferenţa majoră dintre aceste două transformări este că în timp ce k este o marime variabilă, n este o constantă. Într-o transformare politropă sistemul poate primi sau ceda caldură. Noţiunea de transformare politropică este foloşită mai ales în studiul proceselor de comprimare şi de destindere în motoarele cu gaz. Procesele reale de comprimare şi de destindere din motorele cu gaz şi din compresoare nu sunt de obicei nici adiabatice nici izotermice ci ocupă o poziţie intermediară între aceste doua transformări. Din această cauză în practică valorile lui n sunt cuprinse între 1 şi k. Ecuaţia generală a transformării politropice este dedusă după cum urmează:
VdpdHQpdVdUQ
−=+=
δδ
⇒
dpVpHdTCCdT
dVpVUdTCCdT
Tp
TV
−
∂∂
+=
+
∂∂
+=
: dT la p = ct ⇒
pTVp
TV
TVp
VUCC
dTdVp
VUCC
∂∂
+
∂∂
=−
+
∂∂
=−
⇒ dVVT
CCCCdT
TVdT
dVCC
CCpvp
V
p
vp
V
∂∂
⋅−
−=⇒
∂∂
⋅=−
− 1
Se notează echivalentsau V
p
V
Vp
CCCC
nCCCC
n−
1−
=−−
=− şi rezultă:
( ) 0dar 1 =
∂∂
+
∂∂
⇒
∂∂
+
∂∂
=
∂∂
−= dVVTndp
pTdV
VTdp
pTdT
TVdVndT
pVpV
p
Deci ecuaţia procesului politropic este:
0=
∂∂
+
∂∂ dV
VTndp
pT
pV
5.34
Ecuaţia este aplicabilă oricărui sistem termodinamic. Aplicaţie pentru gazul ideal.
10
1o. Procesul adiabatic.
ctpV VdV
Vdp
pR
dV
VR
dp
TVdV
TpdpnC
pV
==+⇒=+⇒
∂∂
+
∂∂
⇒=⇒= γγνγνγγ deci si 000
2o. Procesul izoterm.
ctpV
pR
dV
VR
dpnCdT ==+⇒=⇒∞=⇒= deci si 010 νν
3o. Procesul izocor.
∞→= nCC V deci si
•
V
p
V=ctadiab
T=ct.
P=const
Lucrul mecanic de destindere efectuat de către sistem în cursul unei transformări politropice se calculează din integrala:
∫=2
112 pdVL şi pentru că n
n
VVpp 11= rezultă:
11
sau 11
1
1
211
1
2
11112
−
−=
−
−=
−− n
12
n
pp
nVpL
VV
nVpL 5.35
pentru gazul perfect se obţin ecuaţiile:
TT-1
1sau
1sau 1
1 1
2112211
1
2
1112
−
=−−
=
−
−=
−
nVpL
nVpVpL
VV
nRTL 1212
n
5.36
• Căldura furnizată sau primită de sistem se determina pornind de la principiul I al
termodinamicii:
Q21 = (U2 – U1) + L1-2 5.37
∫
−
∂∂
+∫=−
⇒
∂∂
+
∂∂
=+=
21
2112
,
VV
V
TT V
TV
dVpTpTdTCUU
dVVUdT
TUdUpdVdUQδ
5.38
dacă transformarea politropă este efectuată de un gaz ideal (aproximaţia este valabilă în calculele inginereşti din transformările politrope ale motoarelor cu gaz)
11
( )1
;12122
1 −∆
−=−==− ∫ nTRLTTCdTCUU 12V
T
T Vν 5.39
Să analizăm dependenţa căldurii molare politrope pentru gazul perfect de indicele politropei;
( )VVV C
1nnC
nCC
nRCC
−−
=−
−+=
−+=
γγ ; 1
1C ; 1
V 5.40
Fig. Variaţia căldurii molare politrope, C, în funcţie de indicele politropei
C C→ C
V CV C→CV
1 γ n 1<n<γ izotermă
p
C→C
∞
Zona haşurată corespunde proceselor politropice caracteristice maşinilor termice. Raportul Q/L se poate interpreta pentru toate transformările în cazul gazului ideal.
( ) ( )( )
( )1
; 1
11-n
;1−−
=−⋅
−
−=
∆−∆
=γγ
γ
γν
ν nLQnC
CnLQ
TRnTC
LQ
V
V 5.41
Avem cazurile: Dacă n = 0 sau 0 < n < 1 atunci raportul Q/L >1 Dacă n = 1 atunci Q = L Dacă 1 < n < γ atunci raportul Q/L < 1 şi deci gazul consumă căldură pentru a efectua lucru mecanic. Dacă n = γ atunci Q = 0.
•
Dacă n > γ atunci Q/L < 0.
Variaţia entropiei, adică diferenţa dintre entropiile a două stări din cursul transformării politrope este:
∫
∂∂
=− 2
112
T
Tn
dTTSSS 5.42
deoarece este evident că: TC
TS
n
ν=
∂∂ ecuaţia 5.42 capătă forma:
∫=− 2
112
T
TdT
TCSS 5.43
Când C pentru transformarea politropă este constantă atunci variaţia de entropie este:
12
1
212 T
ln TCSS =− 5.44
Se vede că în transformarea politropică entropia variază logaritmic în funcţie de temperatură. Pentru a putea determina valoarea exponentului politropic al unei transformări concrete a unui gaz trebuie să dispunem de date experimentale legate de respectiva transformare. Procedeul cel mai comod consta în foloşirea curbei transformării în diagrama (p,V). Se logaritmează ecuaţia politropei şi se obţine:
12
21
loglogloglog
VVppn
−−
= 5.45
În scara logaritmica curba politropei devine dreapta iar n este panta acestei drepte.
2
1
Log.V
Log p
5.3.3. Procese izoterme
Fig.5.5.
Pentru gazul perfect transformarea izotermă este descrisă de legea Boyle-Mariotte: pV=const., adică la temperatură constantă volumul variază în funcţie de preşiune după o hiperbolă. Izotermele gazelor reale şi ale corpurilor lichide şi solide au forme mai complicate. Vezi Fig. 5.5 pentru gazul real.
Este important să amintim că derivata Tp
V
∂∂ care este de fapt coeficientul de
compreşibilitate izotermă nu poate fi pozitivă pentru nici-o substanţă şi deci creşterea preşiunii după o izotermă determină o scădere a volumului. • Lucrul mecanic de destindere al unui sistem într-o transformare izotermă este
determinat cu relaţia generală:
∫=−2
121
p
ppdVL 5.46
Pentru calculul integralei este necesar să se cunoască variaţia preşiunii în funcţie de volum de pe izotermă fie din ecuatia de stare fie direct din date experimentale în acest ultim caz integrala calculându-se prin metode numerice.
13
Pentru gazul perfect, L se obţine cunoscând ecuaţia termică de stare:
1
221 ln
VVRTL =− 5.47
• Caldura, furnizată sistemului sau cedată de acesta într-o transformare izotermă se calculeaza cu ajutorul relaţiei:
TdS = δQ ⇒ Q1-2 = T (S2 – S1) 5.48 unde S1 şi S2 sunt entropiile în cele doua stări. Dacă gazul este perfect, atunci din principiul I se ştie că în transformarea izotermă lucrul mecanic efectuat de gaz este egal cu cantitatea de căldura primită:
QL = • Variaţia de entropie într-o transformare izotermă se calculeaza astfel:
( ) ( ) ∫
∂∂
=− 2
1
,, 1122
p
pT
dppSTpSTpS 5.49
şi ţinând cont de ecuaţia lui Maxwell:
∫
∂∂
=− 2
112
p
pp
dpTVSS 5.50
Dacă se cunosc volumele din starile 1 şi 2 atunci relaţia:
( ) ( ) ∫
∂∂
=− 2
1
,, 1122
V
VT
dVVSTVSTVS 5.51
şi ţinând cont de ecuaţia lui Maxwell:
∫
∂∂
=− 2
112
V
VV
dVTpSS 5.52
Pentru calcularea integralelor care intervin în ec (5.50, 5.52) este necesar să se cunoască valorile derivatelor parţiale ale substanţelor studiate pe izotermă. Aceste cantităţi pot fi calculate fie cu ajutorul ecuaţiilor de stare fie prin diferenţierea numerică sau grafică a datelor experimentale asupra relaţiilor existente între p, V şi T. În cazul gazului perfect avem:
pR
TV
VR
Tp
pV
νν=
∂∂
=
∂∂ ; 5.53
şi ca urmare:
1
212
2
112 ln ;ln
VVRSS
ppRSS νν =−=− 5.54
• Căldura molară într-o transformare izotermă este infinit de mare : ∞→TCadică sistemul este un termostat. 5.3.4. Procese izobarice Parametri de stare în transformarea izobară sunt legaţi prin ecuaţia care se deduce pornind de la transformarea politropă care are indicele n=0:
14
1
1
2
2
TV
TV
= 5.55
De aici se vede că pentru gaz cu cât temperatura sa este mai ridicată cu atât este mai mare volumul său. Pentru gaze reale, lichide şi solide încălzirea este însoţită de o dilatare termică de-a lungul izobarei. Aceeaşi creştere a temperaturii produce o dilatare mai accentuată unui gaz decât unui lichid sau unui solid. • Lucrul mecanic de destindere al unui sistem într-o transformare izobară este dat
de relaţia: 5.56 ∫=
2
112 pdVL
Pentru gaz perfect, având ecuaţia de stare: pV = νRT
( )1212 TTRL −=ν 5.57 • Căldura furnizată sistemului în timpul încălzirii sale sau cedată de sistem în timpul răcirii sale, într-o transformare izobară va fi deteminată, plecând de la principiul I: δQ = dU + pdV sau δQ = dH – Vdp
Q1-2 = H2(p,T2) – H1(p,T1) 5.58 Valorile entalpiei pentru cele doua stări pot fi aflate fie din tabelele cu proprietăţi termodinamice ale substanţelor fie cu ajutorul diagramelor de stare ale acestora. Diferenţa dintre entalpiile a două stări de pe izobară poate fi exprimată la rândul sau după cum urmează:
( ) ( ) ∫
∂∂
=− 2
11122 ,,
T
Tp
dTTHTpHTpH 5.59
şi ţinând seama de expreşia căldurii molare la preşiune constantă
5.60 ( ) ( ) ∫=− 2
11122 ,,
T
T pdTCTpHTpH ν
sau ∫=−2
112
T
T pdTCQ ν
Dacă Cp este independentă de temperatură, atunci relaţia devine:
Q2-1 = νCp(T2 – T1) 5.61 • Variaţia de entropie într-o transformare izobară este dată de relaţia:
( ) ( ) ∫
∂∂
=− 2
11122 ,,
T
Tp
dTTSTpSTpS 5.62
sau ( ) ( ) ∫=− 2
11122 ,,
T
T
p dTTC
TpSTpSν
5.63
În cazul în care căldura specifică nu depinde de temperatură în intervalul de temperatură conşiderat, se obţine:
( ) ( )1
21122 ln,,
TTCTpSTpS p=− 5.64
Deci se observă că în transformarea izobară entropia variază în funcţie de temperatură într-un mod logaritmic. 5.3.5. Procese izocore
15
Forma generală a izocorelor unui gaz real într-o, diagrama (p,T) şi (p,V) este reprezentata în fig .
Parametri într-o stare pot fi determinati cunoscând parametri într-o stare de referinţă şi foloşind diagramele de stare, ecuaţiile caracteristice şi tabelele cu proprietăţile termodinamice ale substanţei studiate. Parametri de stare pentru gazul ideal sunt legaţi prin ecuaţia care se poate determina şi dintr-o politropă cu n tinzând la infinit. Creşterea temperaturii gazului care se află într-un volum constant determină totdeauna creşterea preşiunii iar această creştere este cu atât mai rapidă cu cât volumul este mai mic. Încălzirea gazelor reale şi a lichidelor provoacă tot o creştere a preşiunii; mai mult, această creştere este senşibil mai rapidă în lichide decât în gaze. Este interesant de remarcat o particularitate curioasă a izocorelor apei la temperaturi scăzute. Se ştie că la temperatura de 3,98oC denşitatea apei este maximă la preşiune atmosferică normală. Un studiu detaliat arată că în acest domeniu al temperaturii izocorele apei sunt de forma reprezentata în Fig. 5.7; izocorele cu v<1 000 000 ml/g trec prin minim în vecinatatea punctului cu temperatura 3,98 0C, iar izocora v=1000 000 ml/g este tangentă la curba de saturaţie. La stânga punctelor de minim izocorele
au panta negativă, adică VT
p
∂∂
<0.
Fig. 5.7. Izocorele apei pentru v>1, 000 000 ml/g pâna la valoarea v=1, 000 132 ml/g, care corespunde punctului triplu, au particularitatea că intersectează de doua ori curba de saturaţie: o dată cu panta pozitiva iar o data cu panta negativa. Astfel, dacă apa parcurge o transformare izocoră pentru T<TA,încalzirea sistemului duce la micşorarea preşiunii. Când temperatura creşte sistemul care corespunde izotermelor 1,000000 ml/g<v<1, 000 132 ml/g trece de la starea cu o şingura faza la starea cu doua faze, iar apoi din nou la starea cu o şingură fază. • Lucrul mecanic de destindere într-o transformare izocoră este zero. Aceasta se vede din relaţia :
16
∫=2
112 pdVL = 0 5.65
dar şi din L pentru transformarea politropică. • Caldura furnizată sistemului într-o încălzire izocoră se calculează pornind de la principiul I: δQ = dU + pdV ; δQ = dU şi în consecinţă expreşia ei va fi:
Q21 = U2(V,T2) – U1(V,T1) 5.66 Diferenţa dintre energiile interne ale celor două stări pentru transformarea izocoră se determină din:
5.67 ( ) ( ) ∫=− 2
11122 ,,
T
T V dTCTVUTVU ν
Deci căldura se determină ca integrală din:
∫=−2
112
T
T V dTCQ ν
Dacă se foloseşte căldura molară sau specifică medie atunci: Q2-1 = νCV
med(T2 – T1) 5.68 iar dacă căldura molară este constantă ca în cazul gazului ideal expreşia devine: Q2-1 = νCV(T2 – T1) • Variaţia entropiei într-o transformare izocoră se determină astfel:
( ) ( ) ∫
∂∂
=− 2
11122 ,,
T
TV
dTTSTVSTVS 5.69
şi ţinând seama de relaţia Maxwell:
( ) ( ) ∫=− 211122 ,, T
TV dT
TC
TVSTVSν
5.70
Dacă în intervalul specificat căldura specifică este constantă atunci:
( ) ( )1
21122 ln,,
TTCTVSTVS Vν=− 5.71
deci de-a lungul izocorei entropia variază logaritmic cu temperatura. 5.4. Laminarea. Efectul Joule -Thomson Se stie din experienta, ca daca un jet de gaz sau de lichid care curge de-a lungul unui tub sau al unei conducte, întâlneste un obstacol care produce o micsorare brusca a sectiunii transversale a jetului, iar în continuare sectiunea jetului creste, atunci preşiunea gazului (lichidului) este totdeauna mai mica dupa obstacol decât înainte de el. Un asemenea obstacol se numeste rezistenta locala. Efectul micsorarii preşiunii jetului de fluid motor care curge printr-o gâtuire a unei conducte poarta numele de laminare. Laminarea este un proces frecvent în practica: curgerea apei printr-un robinet partial închis sau trecerea aerului printr-un registru partial deschis. Efectul produs de dispozitivele de gâtuire se manifesta prin micsorarea preşiunii dupa rezistenta locala. Din punct de vedere fizic, scaderea preşiunii dupa o rezistenta locala se explica prin dişiparea fluxului de energie pentru învingerea rezistentei locale.
17
Studiem curgerea unui gaz (lichid) de-a lungul unui tub care prezinta o rezistenta locala, de exemplu o diafragma.
Înainte şi după diafragmă secţiunea tubului este aceeaşi şi astfel vom neglija variaţia energiei cinetice a fluidului în curgere. În timpul laminarii fluidul motor poate primi caldură, dar noi vom studia cazul când acest proces este adiabatic, adică curgerea fluidului prin diafragmă se face fără schimb de căldură (pereţii tubului vor fi într-un înveliş adiabatic). Să conşiderăm la un moment dat o masă de gaz cuprinsă între cele două secţiuni, I înainte de rezistenţa locală şi II după rezistenţa locală, de fapt două pistoane imponderabile (Fig.5.8.). Deoarece gazul curge este normal ca cele două pistoane să se deplaseze de-a lungul tubului. Notam cu ∑ aria secţiunii tubului adică aria pistoanelor convenţionale şi cu p1, V1, T1 şi p2, V2, T2 presiunile, volumele şi respectiv temperaturile gazului înainte şi după diafragmă. Într-un anumit interval de timp pistonul I se va deplasa pe o distanta l1 iar pistonul II pe o distanta l2>l1 deoarece presiunea şi densitatea gazului sunt mai mici dupa diafragma decât înaintea ei. Lucrul mecanic efectuat pentru deplasarea pistonului pe distanta l1 va fi LI=p1l1∑= p1V1 cu V1 volumul de gaz împins de pistonul I prin diafragma. Lucrul mecanic efectuat de pistonul II care se deplaseaza împotriva presiunii p2 va fi LII=p2V2. Prin deplasarea masei de gaz considerate se produce într-un interval de timp dat un lucru mecanic egal cu diferenta LII - LI = p2V2 - p1V1. Acest lucru mecanic este consumat pentru învingerea rezistenţei locale; el se transformă în căldură. Deoarece procesul examinat este adiabatic lucrul mecanic este rezultatul variatiei energiei interne: L = -(U2 - U1) Deci: p2V2 - p1V1 = -(U2 - U1) şi U 2 = U1 + p1V1 2 + p2V
Adică H1 = H2 5.72 Rezultatul obţinut este important aratând că: în urma laminării adiabatice valorile entalpiei unui fluid sunt aceleaşi înainte şi după rezistenţa locală. Ne interesează cum variază alţi parametri sau funcţii în acest proces de laminare care este un proces ireversibil. Într-adevăr, dacă imaginăm procesul în sens invers (în tub se inverseaza sensul de curgere) totdeauna se constată o micşorare a presiunii după trecerea gazului prin rezistenţa locală. Acest fapt este natural deoarece energia fluxului este consumată pentru a învinge rezistenţa locală atât într-un sens cât şi în celălalt.
18
Întrucât transformarea este adiabatică şi ireversibilă, entropia gazului (lichidului), creşte. Înainte de a calcula entropia trebuie facută următoarea observaţie: ecuaţiile diferenţiale ale termodinamicii folosite pentru calcularea variatiilor temperaturii, entropiei ,etc sunt sunt aplicabile transformarilor reversibile. Deci pentru a le aplica pentru calculul variatiilor starii gazului în cazul laminarii adiabatice ireversibile din starea 1 în starea 2, trebuie aleasa schema unei transformari reversibile care sa faca sa treaca fluidul considerat din aceeaşi stare 1 (dinainte de diafragma) în aceeaşi stare finala 2 (de după). Ca transformare reversibila se poate lua de exemplu o dilatare a gazului cu cedare (primire ) de caldură, astfel încât entalpia gazului să ramână constantă: spre deosebire de laminarea adiabatică ireversibilă, dilatarea izentalpică (dp<0) nu poate fi în acelaşi timp şi adiabatică; din δQ = dH - Vdp se vede că pentru δQ=0 şi dp≠0 entalpia nu poate ramâne neschimbată, dH≠0. Dacă se imaginează o transformare reversibilă adiabatică şi izentalpică atunci gazul nu va suferi nici-o dilatare sau altfel spus starea gazului nu se va schimba şi deci nu va avea loc nici-o transformare. Variaţia entropiei gazului care rezultă din această transformare reversibilă (variatie care este egala cu cea a entropiei în laminarea gazului din starea 1 în starea 2) va fi:
S ) - S1(H, p1) = ∂∂Sp
dpp
p
H
∫
1
2
5.73 2 (H, p2
care se poate scrie, ţinând seama de relaţia fundamentală:TdS VdpdH −=
∫−=−2
11122 ),(),(
p
pdp
TVpHSpHS 5.74
sau
∫=−1
21122 ),(),(
p
pdp
TVpHSpHS 5.75
aceasta ultimă expresie este mai comoda pentru ca p2< p1, şi din ecuaţie rezultă că întotdeauna S2>S1, deci entropia creşte. Să examinăm acum variaţia temperaturii gazului sau lichidului în cursul laminării adiabatice. Întrucât această transformare este caracterizată prin condiţia H = const. pentru rezolvarea acestei probleme este necesar sa calculăm valoarea derivatei
∂∂TP H
Din relaţia de ciclicitate: 1−=
∂∂
⋅
∂∂
⋅
∂∂
pTH TH
Hp
pT
⇒
p
T
H
THpH
pT
∂∂
∂∂
−=
∂∂
Pentru a evalua expresia Tp
H
∂∂ se porneşte de la relaţia fundamentală a
termodinamicii:
19
pVpH
TT
TH
TS
TTHp
pHH
TpHH
pTVH
TS
Tp
pT
ddd
ddd
ddd
−
∂∂
+
∂∂
=⇒
∂∂
+
∂∂
=
=
−=11),(
1
),( TpSS = ⇒ ppST
TSS
Tp
ddd
∂∂
+
∂∂
= ,
şi identificând termenii celor două ecuatii se obţine:
pp TH
TTS
∂∂
=
∂∂ 1 ş i
−
∂∂
=
∂∂ V
pH
TpS
TT
1 . 5.76
pTS
TpS
∂∂∂
=∂∂
∂ 22
⇒ VTVT
pH
pT
+
∂∂
−=
∂∂ .
p
p
H C
VTVT
pT
−
∂∂
=
∂∂ . 5.77
Marimea ∂∂TP H
se numeste coeficient de laminare adiabatică sau efect Joule-
Thomson diferenţial şi se noteaza cu αH. În cazul general αH este diferit de zero. Variaţia temperaturii gazelor şi lichidelor ca urmare a laminării adiabatice a primit numele de efect Joule - Thomson. Marimea αH se numeşte coeficient Joule - Thomson. Măsurând efectul Joule - Thomson diferenţial, adică o diferenţă finită de temperatură ∆T foarte mică, pentru o diferenţă de presiune de acelaşi ordin de marime de-o parte şi de alta a diafragmei, se poate determina αH şi apoi cunoscându-l pe αH se poate construi diagrama (H, T) a substanţei de studiat, se pot determina caldura specifică, volumul specific şi alte funcţii calorice. Variaţia temperaturii gazului (sau lichidului) în procesul de laminare adiabatică, în cazul unei schimbări însemnate a presiunii în zona de rezistenţă, a primit numele de efect Joule-Thomson integral şi este descris de relaţia:
5.78 ∫=−2
1
p
p
dpTT Hα12
în care T1şi T2 sunt temperaturile fluidului înainte şi dupa diafragmă. Efectul Joule Thomson integral poate atinge o valoare foarte mare. De exemplu, în laminarea adiabatică a vaporilor de apă de la 29 400 kPa la 450oC pâna la 98 kPa, temperatura vaporilor scade până la 180 oC, adică cu 270 oC. Se pune urmatoarea întrebare: temperatura fluidului scade întotdeauna într-o laminare adiabatică sau care este semnul efectului Joule- Thomson? Pentru că întotdeauna Cp > 0, semnul efectului Joule-Thomson diferenţial αH este dat de semnul expresiei:
−
∂∂ V
TVT
p
5.79
20
este evident că dacă
TV
TV
p
<
∂∂ 5.80
atunci αH < 0 iar laminarea adiabatică este însoţită de creşterea temperaturii fluidului.
Dacă TV
TV
p
>
∂∂ 5.81
αH > 0 iar laminarea adiabatică duce la micşorarea temperaturii fluidului.
În sfârşit, dacă TV
TV
p
=
∂∂
5.82
atunci αH = 0 adică temperatura fluidului ramâne neschimbată în laminarea adiabatică. Relaţia este adevarată pentru gazul perfect, ceea ce înseamnă că în laminare gazul perfect nu îşi modifica temperatura. Deci efectul Joule-Thomson nu se manifestă decât în gazele reale ( şi pentru un gaz care se supune ec. Van der Waals) şi
în lichide. Fig.5.9. Curba de inversie a azotului în diagrama (p, T) Experienţa arată că pentru aceeaşi substanţă semnul lui αH depinde de domeniul stărilor acestei substanţe. Starea fluidului în care αH este nul a căpătat numele de punct de inversie al efectului Joule - Thomson. locul geometric al punctelor de inversie într-o diagramă de stare a unei substanţe se numeste curba de inversie a efectului Joule - Thomson. În Fig. 5.9 este reprezentata curba de inversie a azotului în diagrama (p, T). În interiorul regiunii delimitate de curba de inversie αH> 0, adică laminarea antrenează o răcire a gazului. În exteriorul curbei αH <0 adică procesul este însotit de o încalzire a gazului. Curbele de inverşie a altor substante au o alura şimilara. Laminarea adiabatica poate fi foloşita ca un procedeu eficient pentru racirea gazelor. Este clar ca un gaz nu se va raci decât în cazul în care starea sa se va afla în regiunea în care αH>0 adica în regiunea şituata sub curba de inverşie. Este interesant sa comparam acest procedeu de racire cu cel prin destindere adiabatica reversibilă despre care am vorbit într-un paragraf precedent. Vom compara pentru
aceasta valorile derivatelor: ∂∂
∂∂
Tp
si TpH S
.
21
Ultima derivată poate fi numită coeficient de dilatare adiabatică reversibilă (adica izentropică) şi prin analogie cu αH o vom nota cu αS şi poate fi reprezentat din ecuaţia de ciclicitate sub forma:
1−=
⋅
⋅
pTS TS
Sp
pT
∂∂
∂∂
∂∂
şi tinând seama de ec Maxwell, se obţine :
α
∂∂
Sp
p
T VT
C=
5.82
potrivit ecuaţiei 5.82 coeficientul de laminare adiabatică (adică de dilatare adiabatică reversibilă) este
p
pH C
VTVT −
=∂∂
α
şi se obţine: α αS Hp
VC
− = 5.83
Deoarece valorile lui V şi Cp sunt pozitive, αS > αH Ca urmare, se vede ca dilatarea adiabatică reversibilă (cu lucru mecanic exterior) asigură din punct de vedere termodinamic, o răcire mai eficientă a unui gaz sau a unui lichid decât laminarea adiabatică, adică o destindere adiabatică ireversibilă. 5.5. Destinderea adiabatică a unui gaz real în vid (detenta Joule) Sa examinăm încă o transformare ireversibilă tipică unui gaz real, dilatarea adiabatică în vid, fără lucru mecanic exterior. În cazul unui gaz perfect, studiul experimental al dilatării adiabatice fără lucru mecanic exterior a permis lui Gay- Lussac şi apoi lui Joule să stabilească că temperatura gazului ideal nu variaza într-o astfel de transformare; acest fapt a permis să se enunţe o proprietate importantă a gazelor perfecte: energia internă nu depinde de volum. Să studiem legile destinderii adiabatice a gazului real în vid, pornind de la experimentul care presupune că şi la cel al gazului ideal un vas împărţit în două părţi printr-un perete despărţitor mobil; o parte conţine gaz la temperatura T1 în timp ce cealaltă este vidată. Vasul este izolat adiabatic, deci nu există nici-un schimb de căldură cu mediul exterior. La deblocarea peretelui gazul se destinde în vid, determinând o micşorare a presiunii , gazul ocupând întreg volumul V. Temperatura şi entropia variază în aceasta transformare şi variaţiile le vom calcula în cele ce urmează.
22
Primul principiu al termodinamicii se scrie: dVpdUQ mediu+=δ
P2 V2 T2 P1 V1 T1 P=0
Dilatarea este adiabatică iar L =0 deoarece pmediu = 0, gazul destizându-se fără efectuare de lucru mecanic. Ca urmare dU = 0 şi deci U = const., adică în timpul destinderii adiabatice a gazului în vid, energia internă a gazului rămâne neschimbată. Variaţia temperaturii gazului în funcţie de volum într-o transformare U = const. se determină din relaţia:
dVVTVUTVUT
V
V U∫
∂∂
=−2
1
),(),( 1122 5.84
Derivata parţială ∂∂
TV U
care intervine în această relaţie poate fi pusă sub forma:
TVU V
UUT
VT
∂∂
∂∂
−=
∂∂
şi v
V
U CTpTp
VT
∂∂
−=
∂∂ 5.85
şi înlocuind valoarea în ecuatia 5.84 se obţine:
dVC
TpTp
VUTVUTV
V V
V∫
∂∂
−=−
2
1
),(),( 1122 5.86
Această relaţie arată că în cazul general temperatura gazului se micşorează în timpul destinderii adiabatice în vid. Deoarece pentru o substanţă reală avem întotdeauna:
0>−
∂∂
=
∂∂ p
TpT
VU
VU
şi CV>0
în transformarea examinată avem, T2 < T1. Pentru un gaz perfect destinderea adiabatică în vid nu produce nici-o variaţie a temperaturii:
0.
=
∂∂ perf
TVU şi ca urmare
0.
=
∂∂ perf
UVT 5.87
23
Cunoscând valorile V2 şi T2 nu este dificil cu ajutorul diagramelor de stare sau a tabelelor cu proprietăţile termodinamice să se afle valoarea presiunii gazului din vas după dilatarea lui pâna la presiunea p2.. În transformarea studiată variaţia entropiei gazului este dată de relaţia evidentă:
dVVSVUSVUS
V
V U∫
∂∂
=−2
1
),(),( 1122 5.88
şi ţinând seama de relaţia:
Tp
VS
U
=
∂∂ 5.89
se obţine:
( ) ( ) dVTpVUSVU
V
V∫=− 2
11122 ,,S 5.90
integrala din ec (5.90) se calculează cu ajutorul tabelelor proprietăţilor termodinamice ale gazelor (se construieste curba functiei p/T=f(V) pentru U=const, apoi se integrează aceasta curba între V1 şi V2). Întrucât integrala din membrul al doilea al acestei egalităţi este totdeauna pozitivă, avem: S2 > S1., adica în destinderea Joule, care este o transformare ireversibilă tipică, entropia creşte. Pentru un gaz ideal, înlocuind valoarea lui p/T din ecuaţia de stare din relatia (5.90) se obţine:
RTpV =
( ) ( )1
21122 ln,,
VVRVUSVUS =− 5.91
Se constată aceeasi dependenţă de tip logaritmic a variaţiei entropiei pentru gazul ideal. 6. PRINCIPIUL AL III-LEA AL TERMODINAMICII 6.1. Formularea principiului al III-lea al termodinamicii La definirea scării termodinamice a temperaturii s-a subliniat faptul ca ipoteza existenţei unui sistem care ar avea temperatura termodinamica egala cu zero (zero absolut) este în contradicţie cu principiul al II-lea al termodinamicii. În schimb, existenţa unor stări cu temperaturi oricât de apropiate de zero absolut nu contravine nici unui principiu. Are deci sens să ne întrebăm ce devin anumite mărimi termodinamice atunci când temperatura tinde catre zero absolut. Vom studia în special entropia ca functie de T şi V sau T şi p.
∂∂
ν
∂∂
ν
ST
CT
ST
CT
V
V
p
p
= >
= >
0
0 6.1
24
adică entropia este monotonă în raport cu T: descreşte o dată cu descreşterea temperaturii. O teoremă matematică ne asigură atunci, că dacă T→0, entropia tinde către o valoare finită sau -∝ :
∞−
=
∂∂
>→ finitT
S
VTTlim
00
∞−
=
∂∂
>→ finitT
S
pTTlim
00
6.2
Ca urmare putem reprezenta grafic, S(V,T) pentru două valori ale volumului V1şi V2 cu V1> V2.
Fig.6.1. Dependenta entropiei de T când T→0
Fig. 6.1a.contravine principiului al II-lea: dacă efectuăm o comprimare izotermă (A→B) şi apoi o destindere adiabatică(B→C), constatăm că fluidul atinge starea pentru T = 0 înainte ca volumul să fi atins volumul V1, deci contrazice afirmatia: fiecarei stari îi corespunde o anumită entropie. Cerinţele principiului al II-lea nu sunt respectate decât dacă limita entropiei este aceeaşi fie finită sau infinită oricare ar fi volumul (din 6.1b şi 6.1c). În urma unor cercetări foarte ample asupra comportării sistemelor la temperaturi foarte joase, W. Nernst (1906) a ajuns la concluzia că în natură nu poate fi realizată decât starea 6.1c, admisă de principiul al II-lea al termodinamicii. Posibilitatea 6.1b deşi conformă cu principiul al II-lea este interzisă de principiul Nernst. Deci în cazul 6.1c, S = S0 = const., S0 fiind o constantă care nu depinde de parametri de stare. Această relaţie constituie expresia matematică a teoremei Nernst. Teorema Nernst: La zero absolut entropia oricărui sistem termodinamic are o valoare constanta (un proces izoterm este şi izentropic sau izoterma de zero absolut coincide cu adiabata).
25
Analizând concluziile lui Nernst, Planck formuleaza cel de-al III-lea principiu al termodinamicii: în cazul sistemelor omogene condensate, entropia tinde către o valoare limita nulă, când temperatura tinde catre zero absolut.
S = S0 =0 6.3 Formula nu se aplica la sisteme policomponente (solutii, aliaje ) şi solidele amorfe. Altă formulare a Teoremei Nernst-Planck: la zero absolut entropia oricărui sistem termodinamic la echilibru este zero, sau izoterma de zero absolut coincide cu adiabata de zero. 6.2. Consecintele principiului al III-lea 6.2.1. Principiul al III-lea are rolul de a fixa valoarea constantei entropiei . Funcţiile termodinamice se determină pânăla o constanta aditivă. Valorile acestor constante nu pot fi cunoscute pe baza principiilor I, II ; principiul al III-lea determină funcţiile termodinamice ca valori absolute şi nu ca variaţii.
∫=S2
1 TQδ 6.4
starea 1 fiind aleasă arbitrar. sau
∫=T
TQS
0
δ 6.5
Relaţia 6.5 este valabilă dacă în domeniul de temperaturi 0 ÷T nu au loc transformări de stare de agregare sau transformări polimorfe. Dacă astfel de transformări au loc, integrarea se face pe porţiuni şi se însumează entropiile care însoţesc transformările respective. Astfel, de exemplu, la încălzirea unui mol de substanţă solidă care în intervalul de temperatura 0 ÷T trece în stare lichidă şi apoi în stare de vapori, entropia molară la temperatura T şi presiunea de 1 atm va fi :
dTT
CT
dTT
CT
dTT
CS
T
vT
gp
v
vvT
tT
lp
t
ttT
spT ∫++∫++∫= .,
0
, λλ 6.6
6.2.2. Imposibilitatea principială a atingerii temperaturii de zero absolut
Fig. 6.2. Ciclul Carnot
zero absolut care conţine izoterma de
Ciclul Carnot în (T,S) contine izoterma de zero absolut. ∆S = ∆S12 = Q/ T Întrucât Q ≠ 0, T≠∝ şi ∆S = 0 pe întreg ciclul, se obţine un rezultat absurd. Deci temperatura de 0 K nu este accesibilă. Unii autori consideră “inaccesibilitatea temperaturii de zero absolut” ca un enunţ echivalent al principiului al III-lea.
26
Fig. 6.3.
Pentru ca S→ 0 când T→ 0, izocorele de V1şi V2 trec prin origine. Variaţiile de temperatură pentru fiecare treaptă de destindere adiabatică alcătuiesc un şir descrescător format din termeni care tind săse anuleze în apropiere de 0 K. Rezultă, că printr-un numar finit de transformări izoterme temperatura de 0 K nu se poate atinge. 6.2.3. Comportarea coeficientilor termici când T→ 0 K.
pTV
V
∂∂
=1α si
TpS
V
∂∂
=1α 6.7
VT
pp
∂∂
=1β şi
TVS
p
∂∂
=1β 6.8
Cum 0),(lim
)=
→TVS
T , 0limlim
0)==
→→βα
TT 6.9
În stările din vecinătatea stării de zero absolut, substanţele îşi pierd proprietăţile de dilatare, de elasticitate termică, în acord cu experienţa. 6.2.4. Comportarea coeficientilor calorici când T→ 0 K
VV T
STC
∂∂
=ν
şi p
p TST
∂∂
=ν
C 6.10
Prin integrare rezultă :
dTTC
TVST
V∫=0
),(ν
şi dTTC
TpST p∫=0
),(ν
6.11
Integralele trebuie să fie convergente. Entropiile din relaţiile 6.10 şi 6.11 sunt finite numai daca CV , Cp→ 0 când T→ 0 K rezulta
lim ( , )T pC T p
→=
00 lim ( , )
T VC T V→
=0
0
27
6.2.5. Limitele principiului al III-lea a) Aplicarea la un număr restrâns de sisteme implică un grad de generalitate redus faţă de celelalte principii. Se contesta că ar fi principiu pentru că nu a introdus nici-o funcţie de stare. b) Limitarea principiului al III-lea la sistemele condensate implică criterii de valabilitate a modelelor fizice. Astfel, sistemul gaz ideal şi gaz real nu sunt în echilibru termodinamic la 0 K şi ecuaţiile lor de stare nu se aplică pentru T→ 0 K (gazul se degenerează, adică gazul se găseşte în starea de degenerare, fenomen explicat de statistica cuantică).
Exemplu: CV = ct Cp = ct. α β= =1T
S(V,T) = ν dT∫ S(p,T) = C
TV
T
0
νC dTTp
T
0∫
S V T C T R V s
S p T C T R p s
V
p p
( , ) ( ln ln )
( , ) ( ln ln )
= + +
= − +
νν
ν
0
0
V şi ca urmare T→ 0 K implica α β, → ∞→ −∞S
6.3.Limitele de aplicabilitate ale principiilor a) Limita inferioară: nu se aplica la microsisteme. T nu are sens pentru o particulă. Principiul al II-lea stabileşte deosebirea dintre forma microfizica de transmitere a energiei - caldura şi forma macrofizică legată de variaţia parametrilor externi - lucrul mecanic. În cazul sistemelor ale caror dimensiuni sunt comparabile cu dimensiunile moleculare dispare deosebirea dintre notiunile de caldura şi L şi de aceea pentru aceste sisteme (macrosisteme) nu au sens parametrii termodinamici: T, entropie, etc. b) Limita superioară Postulatul termodinamic asupra echilibrului termic, asupra trecerii inevitabile a unui sistem izolat în stare de echilibru şi imposibilitatea de a ieşi de la sine din aceasta stare, precum şi legea creşterii entropiei în asemenea sisteme sunt rezultate ale generalizarii datelor experimentale în sisteme de dimensiuni finite. Extinderea necritică a acestor legi la sisteme de dimensiuni infinite fără analizarea modificarilor profunde poate duce (a şi dus) la concluzii neştiinţifice (moartea termică a universului). Teoria “morţii termice” a fost formulata de Clausius care scria: Energia lumii rămâne constantă, entropia lumii tinde către valoarea maximă , deci mai devreme sau mai târziu Universul ajunge la echilibru termodinamic; atunci toate procesele ar înceta şi lumea s-ar scufunda în stare de moarte termică.
28
7. METODE TERMODINAMICE DE STUDIU
Când se vorbeşte despre metoda termodinamică de studiu a fenomenelor fizice, se are în vedere studiul care se bazează pe folosirea primului şi celui de-al doilea principiu al termodinamicii. Folosirea principiilor termodinamicii pentru rezolvarea unor probleme fizice concrete se face în termodinamica proceselor reversibile prin doua metode: metoda proceselor ciclice (metoda ciclurilor) şi metoda potentialelor termodinamice (sau a funcţiilor caracteristice) 7.1. Metoda proceselor ciclice Metoda proceselor ciclice sau pe scurt metoda ciclurilor are două aspecte: a) Metoda ciclurilor aplicată în vederea obţinerii unor relaţii între diverse mărimi termodinamice care caracterizează un sistem; b) Metoda ciclurilor aplicată la maşinile termice. În primul caz (a), alegerea procesului ciclic de studiu este numai un mijloc metodic care urmăreşte să uşureze obţinerea unor relaţii între diverse mărimi termodinamice ale unui sistem (calduri specifice la gaze reale, presiunea şi temperatura de vaporizare a unui agent termic, etc.). Aceste relaţii se pot obţine şi prin metaoda potenţialelor termodinamice. În al doilea caz (b), însuşi ciclul termodinamic este procesul fundamental care sta la baza funcţionării maşinii termice studiate (motor cu ardere internă, instalaţie de turbine cu gaze, instalaţii frigorifice, etc.). Metoda urmăreşte tocmai studierea acestui ciclu din diverse puncte de vedere: aflarea mărimilor de stare în puncte caracteristice ale ciclului, aflarea lucrului mecanic pe ciclu, determinarea eficienţei ciclului. 7.1.1. Metoda ciclurilor aplicată pentru determinarea unor relaţii fizice. Ideea metodei ciclurilor este următoarea: pentru stabilirea unei legi determinate a unui fenomen se studiază un ciclu reversibil, convenabil ales şi acestui ciclu i se aplică ecuaţia primului principiu al termodinamicii:
δQ L=∫ 7.1
şi ecuaţia celui de-al doilea principiu al termodinamicii: δQ
Trev∫ = 0 7.2
Cu ajutorul acestor ecuaţii se poate ajunge la legea căutată, dacă ciclul este astfel ales încât să existe posibilitatea de a calcula mărimile necesare ce intră în relaţiile 7.1 şi 7.2. Dacă ne imaginăm că sistemul efectuează un ciclu Carnot (cum se face deseori), atunci ecuaţia 7.2. este înlocuită cu expresia randamentului ciclului Carnot. Adică, randamentul ciclului dedus pentru o problemă concretă, se egalează cu
randamentul ciclului Carnot:η = −1 2
1
TT
Din punct de vedere istoric, metoda ciclurilor este cea mai veche metodă de cercetări termodinamice. Carnot, Clausius, Nernst au folosit numai această metodă. Metoda ciclurilor, pe de o parte, principial, poate fi folosită pentru rezolvarea oricărei probleme, dar pe de alta parte are un neajuns destul de mare deoarece pentru stabilirea unei legi, de fiecare dată trebuie ales un ciclu convenabil. Succesul rezolvării problemei depinde aşadar, de alegerea ciclului care nu este determinată în nici-un fel.
1
Aplicatie: Stabilirea cu metoda ciclurilor dependenţa tensiunii superficiale de temperatură.
Fig.7.1
Considerăm un ciclu Carnot efectuat de o peliculă de de lichid pe o ramă de sârmă. În diagrama (σ,Σ) cu σ tensiunea superficială şi Σ suprafaţa peliculei este reprezentat ciclul. Se întinde pelicula izoterm din starea (1) până în starea (2). Din experienţă se ştie că tensiunea superficială nu variază;pentru ca procesul să decurgă izoterm se transmite peliculei căldura Q la temperatura T. Se întinde adiabatic pelicula până în starea (3); în acest caz temperatura scade cu dT, iar tensiunea superficiala se măreşte cu dσ. Apoi se dă posibilitatea peliculei să se comprime, la început izoterm până în starea (4) când cedează căldura Q2 şI apoi adiabatic până în starea (1). În acest ciclu pelicula a efectuat un lucru mecanic L=Q1 - Q2, care pe diagramă este egal cu aria ciclului. Deoarece ciclul este parcurs în sens invers, lucrul mecanic va fi negativ: L d= − −( )Σ Σ2 1 σ
ησ
= = −−L
Qd
Q( )Σ Σ2 1 7.3
pentru că pelicula a efectuat un ciclu Carnot, randamentul va fi:
η =− −
=T T dT
TdTT
( ) 7.4
Ca urmare:
−−
=( )Σ Σ2 1 d
QdTT
σ 7.5
şi rezultă:
−−
= ⇒
= −−
⋅Σ Σ
Σ ΣΣ
2 1
2 1
1Q
d dTT
ddT
QT
σ σ 7.6
dar QrΣ Σ2 1−
= λ , căldura latentă de formare a unităţii de peliculă
Deci, ddT T
rσ λ
= −Σ
7.7
Adică, variaţia tensiunii superficiale la creşterea temperaturii, scade. 7.1.2. Metoda ciclurilor aplicată la maşinile termice
2
a) Ciclurile motoarelor cu combustie internă, cu piston Motoarele cu combustie internă (de tipul cu piston) sunt foarte mult folosite
la: automobile. Asa cum îi spune si numele, motorul cu combustie internă este o maşină termică în care fluidul motor primeşte căldură de la un combustibil care arde chiar în interiorul motorului. În aceste motoare fluidul este format, în prima etapă, din aer sau amestec de aer şi un combustibil uşor inflamabil, iar în a doua etapă, de produsele de combustie ale combustibilului lichid sau gazos (benzină, kerosen, motorină, etc.). În motoarele cu gaz fluidul este supus unui regim de presiune nu prea ridicat, iar temperaturile sunt uşor superioare temperaturii critice, ceea ce îi permite sa fie asimilat, cu o bună aproximaţie, unui gaz perfect; aceasta simplifică mult analiza termodinamică a ciclului motor. Motoarele cu combustie interna au două avantaje importante prin comparaţie cu alte motoare termice: i) sunt mai compacte, deoarece sursa caldă fiind în interiorul motorului nu este nevoie de o suprafaţă mare pentru realizarea schimbului de căldura cu fluidul motor; ii) temperatura fluidului motor nu este limitată superior, deoarece fluidul motor primeşte căldură nu numai prin peretii motorului ci şi datorită degajării căldurii care se produce în fluid. În plus, pereţii cilindrilor şi chiulasei sunt echipaţi cu sisteme de răcire forţată. Lărgirea intervalului de temperaturi permite îmbunătăţirea randamentului termic. Corpul principal al oricărui motor cu piston este cilindrul în care se deplasează un piston legat prin intermediul unui sistem bielă-manivelă de receptorul de lucru mecanic. Cilindrul are orificii închise cu supape, dintre care unul serveşte la aspiraţia fluidului motor (a aerului sau a amestecului combustibil), iar celălalt pentru evacuarea fluidului motor după ce s-a realizat ciclul. Se disting trei tipuri principale de cicluri motoare cu combustie internă cu piston: ciclul Otto (combustie la volum constant, V=const), ciclul Diesel (combustie la presiune constantă, p=const) şi ciclul Trinckler (combustie la volum constant, V=const iar apoi la p=const). Ciclul Otto (dupa numele lui N. Otto, inginer german care a realizat acest ciclu în 1876). Schema unui motor care funcţionează după un ciclu Otto şi diagrama dinamica a acestui motor sunt date în Fig. 7.2. Pistonul P este antrenat într-o mişcare rectilinie alternativă în cilindrul C care are o supapă de aspiratie S.a şi o supapă de evacuare S.e. În cursul transformării 1-1’, pistonul se deplasează producând o scădere a presiunii în cilindru, supapa de admisie S.a se deschide, iar amestecul combustibil care este preparat separat în carburator, este aspirat în cilindru. În ciclul Otto amestecul combustibil este aerul amestecat cu o
3
anumită cantitate de vapori de benzină (sau alt combustibil). Când pistonul ajunge la extremitatea din dreapta, supapa de aspiraţie întrerupe admisia amestecului combustibil în cilindru, iar pistonul începe să comprime amestecul. Prin comprimare presiune creşte (transformarea 1-2). În momentul în care presiunea amestecului combustibil atinge o valoare bine determinată, care corespunde stării 2 de pe ciclu, bujia electrică Bj asigură aprinderea gazului comprimat. Arderea amestecului gazos se produce aproape instantaneu, pistonul nereuşind sa se deplaseze în timpul arderii aşa încât se poate considera fenomenul de ardere ca un proces izocor (2-3). Căldura degajată prin ardere încălzeşte fluidul motor care se află în cilindru şi măreşte presiunea până la o valoare care corespunde punctului 3 de pe diagramă. Sub efectul acestei presiuni pistonul se deplasează spre dreapta, producând un lucru mecanic de destindere care este transmis receptorului de lucru mecanic. În momentul în care pistonul atinge punctul mort din dreapta, un dispozitiv special deschide supapa de evacuare S.e, iar presiunea din cilindru scade la o valoare ceva mai mare decât presiunea atmosferică (transformarea 4-5); în acest timp o parte din gazul ars iese din cilindru. Apoi pistonul se deplasează din nou spre stănga, evacuînd în atmosferă restul de gaz ars. (Din diagrama ciclului se observă că presiunea din cilindru este, în timpul aspiraţiei ceva mai mică şi în cursul evacuării ceva mai mare decăt presiunea atmosferică, datorită rezistenţei aerodinamice a celor două supape şi tubulaturii de admisie şi de evacuare.) Apoi începe un nou ciclu: aspiraţia amestecului combustibil, comprimarea, etc. Se vede că într-un motor care funcţionează după un ciclu Otto, pistonul efectuează patru curse pentru un ciclu; se spune ca motorul este în patru timpi: aspiraţia, compresia, destinderea dupa arderea amestecului gazos, evacuarea gazelor arse în atmosferă. Analiza termodinamică a ciclului Otto este uşor de făcut în ipotezele: ciclul este închis deoarece, deşi la sfârsitul ciclului motor fluidul este evacuat astfel ca la fiecare ciclu este alta porţie de combustibil, combustibilul fiind în cantitate mică prin comparaţie cu aerul putem considera cantitatea de fluid motor constantă. Caldură furnizată fluidului motor de sursa caldă se face în cursul transformării 2-3 iar fluidul motor cedează căldură sursei reci în timpul transformării izocore 4-1. Deoarece comprimarea 1-2 şi destinderea 3-4 se produc în intervale de timp exttrem de scurte, nepermiţând schimb de căldură cu mediul înconjurător, aceste transformări se pot considera adiabatice. Ca urmare lucrul mecanic produs de motor este dat de aria ciclului. Randamentul termic al ciclului Otto. Căldurile primite şi cedată în transformările izocore sunt:
( )231 TTCQ V −= ( )142 TTCQ V −= 7.8 Randamentul ciclului ca randamentul oricărei maşini termice va fi:
( )( )
−
−
−=−−
−=1
111
2
32
1
41
23
14
TT
T
TTT
TTCTTC
V
Vη 7.9
4
Ţinând seama de ecuaţiile transformării adiabatice: şi definiţia
raportului de compresie:
122
111
−− = kk VTVT
2
1
VV
=ε rezultă:
12
1 1−= kT
Tε
7.10
Considerând transformările izocore şi din nou cele adiabatice se găseşte relaţia dintre temperaturi după cum urmează:
R
Ccad racbdIdca cmcdcda ncnfuS
3
3
2
2
Tp
Tp
= şi 1
1
4
4
Tp
Tp
= sau
2
3
2
3
pp
TT
= şi 1
4
1
4
pp
TT
=
Pentru că: şi
rezultă:
kk VpVp 4433 =
kk VpVp 1122 =1
4
2
3
pp
pp
= şi ca
34 TT
andamentul ciclului Carnot devine: TT
=
1
11 −−= kεη
urmare 21
onform ecuatiei (7.11), randamentul termic al cicoeficientul de compresie al fluidului motor în transfortât mai mare, cu cât raportul de compresie este mai me raportul de compresie pentru k=1,35 sunt date de în
Concluzia că o comprimare prealabilă a fluidundamentului motorului este foarte importantă şi este
u ardere internă. Aceasta idee a comprimării prealabileruscă a randamentului termic al motorului a maezvoltarea teoriei motoarelor cu ardere internă. eea aceasta a fost prezentată de S. Carnot înca din 1824 i
omprimarea aerului şi cu ardere la volum constant a fost propusă construit un motor care să funcţioneze după ciclul Rochas.
Prin urmare, pentru îmbunătăţirea randamentuluoeficientul de compresie. În practică, însă nu se pot ari, deoarece duc la o creştere considerabilă a tem
onsecinţă se ajunge la o autoaprindere a amesteculuetonaţii care distrug motorul. În motoarele cu carbompresie nu depăşeşte valori între 7 – 12. Valoarea rae calitatea carburantului: el este cu atât mai mare cule carburantului, caracterizate prin cifra octanică, sunt
Raportul de compresie ε dintr-un ciclu se poaumai amestecul combustibil, ci şi aerul pur şi dacă ilindru la sfârşitul comprimării. Pe acest principiu sumele inginerului german R.Diesel, care a construncţioneze după acest ciclu).
chema unui motor Diesel şi diagrama lui dinamică sun
5
Fig.7.3
7.11
lului Otto depinde numai de marea adiabatică 1-2 şi este cu are.Variaţiile lui η în funcţie
diagrama din Fig. 7.3. lui motor permite ameliorarea valabilă pentru toate motoarele a aerului care duce la mărirea
rcat un mare pas înainte în
ar prima schema a unui motor cu în 1862 de A.Beau de Rochas. Otto
i este avantajos să se mărească obţine valori ale lui ε foarte peraturii şi a presiunii şi în
i combustibil şi ca urmare la uratoare obişnuite raportul de portului de compresie depinde
cât proprietăţlie antidetonante mai bune. te mări daca se comprimă nu se introduce combustibilul în
e bazează ciclul Diesel (după it în 1897 un motor care sa
t prezentate în figura 7.4.
În cursul transformării 1’-1 aerul atmosferic pur este aspirat în cilindrul motorului, în timpul transformarii 1-2 acest aer suferă o comprimare adiabatică până la presiunea p2 (raportul de compresie din motoarele Diesel este mărit în general până la ε =15 sau 16). După aceea aerul comprimat începe să se dilate şi, în acelaşi timp lichidul combustibil (kerosen, motorină) este introdus în cilindru printr-un injector special. Datorită temperaturii ridicate a aerului, picăturile de lichid se aprind iar arderea se produce la presiune constantă, astfel încât gazul se dilată de la V2 la V3 la presiune constantă. Din acest motiv ciclul Disel este numit ciclu cu combustie la presiune constantă. Când admisia lichidului combustibil în cilindrul motorului se termină (starea 3), dilatarea ulterioară a fluidului motor se efectuează dupa adiabata 3-4. În starea 4 supapa de evacuare a cilindrului se deschide, presiunea din cilindru scade până la presiunea atmosferică (după izocora 4-5), iar gazele arse sunt evacuate în atmosferă (dreapta 5-1”). Se vede ca ciclul Diesel este un ciclu în patru timpi. Pentru analiza termodinamica vom considera ciclul Diesel închis efectuat de aerul pur, echivalent ciclului real deschis. În diagrama p,V acest ciclu teoretic este de forma din figura 7.5. Se vede ca ciclul Diesel idealizat este compus din două adiabate, dintr-o izobară în cursul căreia se primeşte caldură şi dintr-o izocoră după care se cedează caldură sursei reci. Vom calcula randamentul ciclului Diesel, considerând că aerul care-i fluidul motor este un gaz ideal cu caldura molară constantă. Definim pe lânga raportul de
compresie ε (ε =2
1
VV ) şi raportul de destindere sau grad de destindere prealabilă
2
3
VV
=ρ .
Din expresia generală a randamentului termic al unui ciclu oarecare, 1
21QQ
−=η
Şi ţinând seama de expresiile căldurii primite în transformarea izobară şi cedată în transformarea izocoră,
( )231 TTCQ p −= şi ( )142 TTCQ V −= 7.12 obţinem:
( )( )23
141TTCTTC
p
V
−−
−=η sau, pentru că raportul γ=V
p
CC
pentru gazul perfect,
6
1
111
2
3
1
4
2
1
−
−−=
TTTT
TT
γη 7.13
Ţinând seama de ecuaţia transformării izobare şi ecuaţiile transformărilor adiabatice:
ρ==2
3
2
3
VV
TT
şi
γγ2211 VpVp = şi pentru că Vγγ
3344 VpVp = 14 V= , 32 pp = , obţinem: γ
=
2
3
1
4
VV
pp 7.14
şi înlocuind valorile presiunilor cu temperaturile din ecuaţia transformării izocore 4-1 obţinem:
γρ=1
4
TT 7.15
şi ca urmare expresia randamentului motorului Diesel:
1
11111 −⋅
−−
⋅−= γ
γ
ερρ
γη 7.16
Ecuaţia 7.16 arată ca randamentul termic al unui ciclu Diesel este cu atât mai mare, cu cât raportul de compresie ε este mai mare (la fel ca şi la ciclul Otto) şi cu cât valoarea lui ρ este mai mică. Într-un ciclu Diesel variaţiile randamentului în funcţie de ε , pentru diferite valori ale lui ρ şi pentru 35,1=γ sunt prezentate în figura 7.6. În diagramă (T,S), ciclul Diesel are forma dată în figura 7.7.Cantitatea de căldură Q1 este dată de aria a-2-3-b-a, cantitatea de căldură Q2 prin aria a-1-4-b-a, iar lucrul mecanic pe ciclu prin aria 1-2-3-4-1. Figurile……. Ciclul cu combustie mixtă sau ciclul Trinckler (după numele inginerului rus Trinckler, care a propus acest ciclu în 1904). Acestui ciclu i se mai spune şi ciclul Sabathe şi este o combinaţie a ciclurilor Otto şi Diesel. Motoarele care functionează după acest ciclu (figura 7.8) au o camera numită camera de precombustie, care comunică cu cilindrul de lucru printr-un canal îngust. În figura 7. 9 Este reprezentat acest ciclu în coordonate (p,V).
7
În cilindrul de lucru aerul suferă o comprimare adiabatică, datorită inertiei unui volant cuplat cu arborele motor. Temperatura pe care o atinge aerul în cursul acestei comprimări este suficientă pentru a produce autoaprinderea lichidului combustibil introdus în camera de precombustie (transformarea 1-2). Forma si amplasarea camerei de combustie asigură un amestec mai bun al combustibilului cu aerul, ceea ce favorizează o ardere rapidă a unei părti din combustibil în volumul restrâns din camera de precombustie (transformarea 2-5). Datorită creşteri presiunii în camera de precombustie, amestecul format din combustibilul nears, aer şi gazul rezultat din ardere ajunge în cilindrul de lucru unde se produce arderea, ceea ce produce deplasarea pistonului de la stânga la dreapta la o presiune aproape constantă (transformarea 5-3). Dupa arderea completă, destinderea produselor de ardere (timpul motor) se face adiabatic (transformarea 3-4), dupa care gazele arse sunt evacuate din cilindru (transformarea 4-1).
Aşadar, în ciclul cu combustie mixtă, căldura Q1 este furnizată mai întâi după o izocoră ( )'
1Q iar apoi după o izobară ( Q ). "1
Spre deosebire de motorul Diesel, motorul cu combustie mixtă nu are nevoie de compresorul de presiune înaltă pentru pulverizarea lichidului combustibil. Combustibilul lichid introdus în camera de precombustie, la o presiune relativ redusă, este pulverizat cu ajutorul uni jet de aer comprimat, care provine de la cilindrul principal. În acelaşi timp, ciclul cu combustie mixtă păstrează o parte dintre avantajele pe care le are ciclul Diesel faţă de ciclul Otto, întrucât faza de ardere se realizează la presiune constantă.
Randamentul ciclului cu combustie mixtă se determină înlocuind în relaţia de definiţie a randamentului maşinii termice căldurile primită şi cedată:
"1
'11 QQQ += cu ( )25
'1 TTCV −=Q şi ( )53
"1 TTC p −=Q 7.17
( 142 TTCQ V − )= 7.18
Ca urmare expresia randamentului va fi:
( )( ) ( )5225
141TTCTTC
TTC
pV
V
−+−−
−=η 7.19
sau
−+
−
−⋅−=
11
11
5
3
2
5
2
5
1
4
2
1
TT
TT
TT
TT
TT
γη 7.20
Ţinând seama de ecuaţiile transformărilor izocoră (4-1), adiabată ( 1-2 şi 3-4):
1
4
1
4
pp
TT
= , , 7.21 γγ2211 VpVp = γγ
3344 VpVp =
şi de V se obţine: 21 V=
8
γ
=
2
3
2
3
1
4
VV
pp
pp 7.22
Relaţia (7.22) se poate scrie sub forma, ţinând seama că 43 pp = (izobara 5-3) şi că (izocora 2-5): 52 VV =
γρλ ⋅=1
4
pp 7.23
unde 2
5
pp
=λ este raportul de compresie în timpul combustiei izocore, iar V
V3=ρ ,
raportul de destindere prealabilă în cursul arderii la presiune constantă. Dacă ţinem seama de ecuaţia (7.22) deducem:
γρλ ⋅=1
4
TT 7.24
În continuare daca ţinem seama de relaţiile scrise pentru transformările izocoră (2-5), izobară (5-3) şi de relaţia dintre temperaturile T1 şi T2:
2
5
2
5
pp
TT
= , ρ==5
3
5
3
TT
VV
, 12
1 1−= γεT
T 7.25
Ca urmare expresia randamentului devine:
( ) ( ) 1
111
11 −⋅−+−
−−= γ
γ
εργλλλρη 7.26
pentru 1=ρ (adica un ciclu fară transformare izobară), ecuaţia (7.26) devine ecuaţia pentru randamentul ciclului Otto, iar pentru 1=λ (ciclu fără transformare izocoră), ecuaţia (7.26) se reduce la expresia pentru ciclul Diesel. Compararea randamentului pentru ciclul cu combustie mixtă cu randamentul pentru ciclurile Otto şi Diesel arată că pentru aceleaşi valori ale raportului de compresie ε
OttomixtcombDiesel ηηη << . 7.27 dar pentru aceleaşi valori ale temperaturii maxime (T3) ale ciclului,
OttomixtacombDiesel ηηη >> . 7.28 Aceste inegalităţi sunt evidente din diagrama (T,S) , figura 7.10; de exemplu relaţia 7.28 rezultă din faptul că pentru toate trei ciclurile căldura Q2 are aceeaşi valoare, exprimată prin aria a-1-4-b-a. pentru valoarea maximă a lucrului mecanic al ciclului în ciclul Diesel (aria 1-2-b-3-4-1), valoarea medie a lucrului mecanic al ciclului cu combustie mixtă (aria 1-2-5-3-4-1) şi valoarea minimă a lucrului mecanic în ciclul Otto (aria 1-2a-3-4-1).
9
Rezultatele pe care le-am obţinut, analizând eficacitatea ciclurilor utilizate în motoarele cu ardere internă, nu sunt valabile decât pentru ciclurile idealizate (teoretice), pentru că nu am ţinut seama de ireversibilitate şi de alţi factori. În ciclurile reale fluidul motor (în primii doi timpi este aerul pur în ciclurile Diesel şi cu combustie mixtă şi amestecul combustibil în ciclul Otto, în timpii următori este aerul şi gazul de combustie), diferă, prin proprietăţile sale de un gaz perfect cu căldură specifică constantă. Frecarea fiind inevitabilă, comprimarea adiabatică şi destinderea adiabatică nu se realizează izentropic ci duc la o creştere a entropiei; răcirea forţată a pereţilor cilindrului duce şi ea la creşterea diferenţei dintre aceste transformări şi transformarea izentropică. Arderea se produce în intervale de timp foarte scurte, dar finite, astfel încât pistonul suferă o mică deplasare în timpul arderii, iar ultima nu este riguros izocoră; diverse mecanisme produc pierderi mecanice, etc. Cele spuse se referă şi la timpul de evacuare, care începe după deschiderea supapei de evacuare. Din aceasta cauză, pentru a trece de la ciclurile termodinamice idealizate la ciclurile reale, este necesar să fie introdus randamentul intern relativ la motor, a cărui valoare este determinată experimental în timpul încercărilor care se fac cu motorul. Unul din inconvenientele majore ale motoarelor cu combustie internă cu piston îl constituie folosirea obligatorie a unui sistem bielă- manivelă si a unui volant, ceea ce face inevitabilă funcţionarea lor discontinuă; acest fapt nu permite concentrarea unei puteri mari într-un singur agregat; toate acestea restrâng domeniul de folosire a motoarelor cu piston. Acest inconvenient este înlăturat în motorul cu combustie internă de un alt tip- turbina cu gaz. Aceasta este caracterizată de un randament ridicat şi prezintă în acelaşi timp toate avantajele unui motor rotativ, adica permite realizarea de puteri mari în instalaţii cu gabarite mici. În prezen turbinele cu gaz sunt folosite în aviaţie, în marină, în tracţiunea feroviară şi în centrale. Motorul cu reactie este o maşina motrice care transformă energia chimică a unui combustibil în energie cinetică a jetului fluid motor (gaz) care se destinde în duze. Acest jet furnizează forţă de tractiune motorului, datorită reactiei fluidului care curge în sens opus celui de deplasare a aparatului care zboară. Motoarele cu reacţie se împart în doua categorii principale: motoarele rachetă şi motoarele cu reacţie aerotermice numite simplu reactoare. Aeronavele a căror propulsie este asigurată de motoare rachetă trebuie să ia la bord atât combustibil, cât şi comburant, adică un corp oxidant (oxigen lichid, ozon, peroxid de hidrogen, acid azotic, etc.) care este necesar pentru ardere. Spre deosebire de rachete, paratele echipate cu motoare cu reactie aerotermice au numai combustibil şi utilizează drept comburant oxigenul din aerul atmosferic. Deci motoarele cu reacţie (reactoarele) nu pot funcţiona decât în atmosfera terestră, pe când motoarele rachetă pot fi folosite atât în atmosfera Pământului cât şi în spatiul cosmic.
10
7.2.Metoda potenţialelor termodinamice (funcţiilor caracteristice) În prezent, aproape în toate cazurile de cercetare termodinamică se foloseşte nu metoda ciclurilor prezentată anterior ci aşa numita metodă a potenţialelor termodinamice. Această metoda a fost creată de Gibbs este o metodă analitică şi se bazează pe ecuaţia fundamentală a termodinamicii:
TdS dU A da
TdS dU L
ii
i≥ +
≥ +
∑δ
7.29
7.2.1.Prez
E
entarea generală a metodei Problema centrală a termodinamicii sistemelor la echilibru este de a găsi valorile parametrilor sistemului într-o stare de echilibru (obţinută prin aplicarea asupra sistemului a anumitor constrângeri) şi studiul naturii acestui echilibru (stabil sau instabil). Aceste stări de echilibru sunt complet determinate prin valorile pe care le iau variabilele de poziţie a1.......an şi valoarea pe care o ia încă o variabilă. Această variabilă este energia, U. Atunci ecuaţiile caracteristice ale sistemului se scriu sub forma:
T T U a a aA A U a a a
n
i i n
==
( , , ,.......... )( , , ......... )
1 2
1 2
7.30
unde T şi Ai reprezintă respectiv temperatura şi variabilele de forţă. O alta formă a ecuaţiilor caracteristice se obţine rezolvând prima ecuatie din (7.30) faţă de U (ceea ce se poate face univoc datorită monotoniei funcţiei U: creşte odată cu creşterea temperaturii) şi substituind rezultatul în cea de-a doua ecuaţie.
U U T a a aA A T a a a
n
i i n
==
( , , ,.......... )( , , ......... )
1 2
1 2
7.31
De obicei nu se cunosc toţi parametrii necesari pentru a calcula valorile acestor funcţii în starea de echilibru. De exemplu, dacă pentru un sistem simplu, U şi V sunt considerate variabile independente atunci nu ştim, în general care sunt valorile lor într-o anumită stare de echilibru a sistemului. Deci nu putem calcula p şi T în această stare de echilibru dacă nu cunoaştem ecuaţiile de stare. Această problemă se poate rezolva plecând direct de la principiile termodinamicii, dar pentru calcule şi pentru dezvoltarea teoriei metoda mult mai practică este cea a funcţiilor caracteristice (potenţialelor termodinamice). Metoda potenţialelor termodinamice constă în utilizarea unor funcţii de stare, numite “funcţii caracteristice” pentru studiul sistemelor termodinamice aflate în diferite condiţii de interacţie cu mediul exterior. Se numesc funcţii caracteristice, funcţiile de stare ale unui sistem termodinamic care împreună cu derivatele lor determină complet proprietăţile sistemului. Se numeşte potenţial termodinamic o funcţie caracteristică a cărei valoare descreşte în timpul evoluţiei spre echilibru a unui sistem termodinamic. De ce se cheamă potenţiale? rÎn mecanică dEp= -δL; şi când sistemul este la echilibru mecanic, energia potenţială are valoarea minimă.
F p= −∇
11
Deci potenţialul termodinamic este de fapt o funcţie criteriu al sensului de desfăşurare a procesului, sau criteriu de echilibru pentru sistemele termodinamice din natură, care sunt sisteme neizolate. Pentru un sistem termodinamic simplu U şi V sunt variabile fundamentale care caracterizează starea de echilibru termodinamic. Aceasta afirmaţie este evidentă dacă scriem ecuaţia fundamentală a termodinamicii: TdS dU pdV= + Ecuaţia fundamentala a termodinamicii scrisă pentru un sistem termodinamic simplu, leagă cinci variabile: trei funcţii (T,S,U) şi doi parametri (p,V) de stare. Rezultă că orice potenţial termodinamic trebuie astfel ales încât să depindă de o variabilă din expresia lui δL şi una din expresia lui δQ. După cum vom vedea în continuare, cunoaşterea a cel puţin a unui potenţial termodinamic ne permite să obţinem atât ecuaţii termice de stare cât şi ecuaţia calorică de stare precum şi proprietăţile termodinamice corespunzătoare acestor ecuaţii. Vom începe discuţia cu cea mai importantă funcţie de stare care este funcţie caracteristică dar nu potenţial termodinamic, entropia. 7.2.2. Entropia ca funcţie caracteristică Entropia este funcţie caracteristică dar nu şi potenţial termodinamic pentru că ea creşte şi atinge valoarea maximă când sistemul este la echilibru termodinamic. Este însă cea mai complexă funcţie caracteristică.
Din ecuaţia fundamentală a termodinamicii, dSdU A da
T
i ii=
+ ∑ se sugerează
alegerea variabilelor U şi ai ca variabile independente pentru precizarea stărilor de echilibru. Cu o astfel de alegere, S=S(U, a1,.........an) se numeste ecuaţie entropică fundamentală sau ecuaţie caracteristică generală în reprezentarea entropică deoarece fiind cunoscută, din ea se pot deduce prin derivare ecuaţiile caracteristice. Această ecuaţie se scrie sub forma diferenţială:
dS SU
dU Sa
daa ii
n
Ui
i
=
+
∑∂
∂∂∂
7.32
Comparând cele două ecuaţii se obţine: 1T
SU ai
=
∂∂
şi jaiaUi
i
aS
TA
≠
=
,∂∂ 7.33
sau pentru sistem termodinamic simplu: 1T
SU V
=
∂∂
şi UV
STp
=
∂∂ 7.34
care reprezintă ecuaţia calorică şi respectiv ecuaţia termică de stare. Ele se mai pot scrie şi ca:
T US V
=
∂∂
şi
V
U
USVS
p
=
∂∂∂∂
7.35
12
Ecuaţiile 7.35 sunt echivalente cu ecuaţiile caracteristice (7.30), pentru că permit exprimarea temperaturii şi variabilelor de forţă în funcţie de variabilele independente U şi V. Un sistem termodinamic izolat este definit prin constanţa energiei sale interne (U=const) şi a volumului pe care îl ocupă (V=const). Din ecuaţia fundamentala a termodinamicii pentru procesele nestatice TdS dU pdV≥ + rezultă criteriul: dS ≥ 0; semnul egal corespunde transformărilor reversibile. Prin urmare, la echilibru S este maximă şi se poate scrie:
dS = 0 S = Smax d2S < 0 7.36 Deci, daca un sistem este complet izolat, are loc un proces spontan de trecere de la o stare la alta, numai dacă entropia sistemului creste, adică Sf > Si, pentru U = const. Starea finală de echilibru stabil este cea în care S este maximă. O asemenea mărime poartă numele de funcţie Massieu. Vom considera în cele ce urmează situaţii practice în care sistemul termodinamic este supus anumitor constrângeri şi vom analiza trecerea sistemului dintr-o stare iniţială de echilibru într-o stare finală care poate de asemenea fi o stare de echilibru stabil. 7.2.3. Sistem termodinamic izolat adiabatic: U(S,V) şi H(p,S) În condiţiile în care sistemul este izolat adiabatic, considerând un proces infinitezimal care duce sistemul într-o stare învecinată stării iniţiale, ecuaţia
fundamentală a termodinamicii dST
dU pT
dV≥ +1 se scrie:
dU TdS pdV≤ − 7.37 • Sistemul în condiţii izentropice şi la volum constant Din ecuaţia 7.37 se vede că energia este funcţie de entropie şi volum şi ecuaţia este o ecuaţie caracteristică fundamentală pentru că din ea se pot deduce ecuaţia de stare şi ecuaţia calorică. Într-adevăr, pentru procese reversibile scriind ecuaţia diferenţiala a energiei si comparând-o cu ecuaţia (7.37) obţinem cele două ecuaţii de stare.
dU U
SdS U
VdV
dU TdS pdVV S
=
+
= +
∂∂
∂∂
T US V
=
∂∂
şi p UV S
= −
∂∂
7.38
ecuaţia calorică de stare şi respectiv ecuaţia termică de stare. Parametrii T şi p sunt parametri complementari parametrilor independenţi S şi V şi se vede că reprezintă derivatele de ordinul I ale energiei în raport cu parametri independenţi. Dacă procesul este ireversibil, atunci integrând ecuaţia (7.37) între cele două stări iniţială şi finală rezultă:
U ( )U TdS pdVf ii
f
− < −∫şi pentru că sistemul este izolat adiabatic si este impusă condiţia suplimentară ca volumul să fie constant ( condiţii greu de regăsit în practică), se obţine:
U Uf i− < 0 7.39 Deci U trebuie să fie minima într-o stare de echilibru, în condiţiile impuse.
13
La echilibru energia internă este minimă: U = Umin, dU = 0 d2U > 0 7.40
Prin analogie cu mecanica unde unde echilibrul stabil al unui sistem presupune un minim al energiei potenţiale, funcţia caracteristică U, se numeşte potenţial termodinamic. Cunoscând ecuaţiile caracteristice (7.38) se pot determina funcţiile de răspuns ale sistemului; proprietăţile termice şi calorice ale sistemului termodinamic se determină din derivatele de ordinul I şi II ale funcţiei caracteristice. Astfel, deoarece functia caracteristica are diferenţială totală exactă, din condiţia ∂∂ ∂
∂∂ ∂
2 2UV S
US V
= şi ţinând seama de ecuaţiile (7.38) se obţine:
∂∂
∂∂
∂∂
∂∂V
US S
UV
=
adică relaţia Maxwell:
VS Sp
VT
−=
∂∂
∂∂ 7.41
Derivând ecuaţia termică în raport cu V şi ecuaţia calorică în raport cu S se obţine:
2
2
SU
ST
V ∂∂
=
∂∂ 2
2
VU
Vp
S ∂∂
=
−
∂∂
şi deci:
C TU
S
V =
ν ∂∂
12
2
7.42
şi KV U
V
S = ⋅
1 12
2
∂∂
7.43
• Sistemul în condiţii izentropice şi la presiune constantă. Considerând din nou un proces natural infinitezimal, parametrii independenţi fiind S şi p, ecuaţia fundamentală se scrie:
dU TdS pdV≤ − sau d U pV TdS Vdp( )+ ≤ + 7.44 Cum U+pV =H, entalpia, ecuaţia 7.44 se scrie pentru procese reversibile:
dH TdS Vdp= + 7.45 Entalpia fiind funcţie de stare care depinde de (S,p) diferenţiala ei este diferenţiala totală:
dH HS
dS Hp
dpp S
=
+
∂∂
∂∂
7.46
Comparând ecuaţiile (7.45) şi (7.46), se determină ecuaţia calorică şi ecuaţia termică de stare:
T HS p
=
∂∂
şi V 7.47 Hp S
=
∂∂
T şi p sunt parametri complementari corespunzători parametrilor S şi respectiv V. Ecuaţia calorica de stare sau ecuaţia Gibbs-Helmholtz se obţine imediat din definiţia entalpiei şi sub forma:
14
U H pV H p Hp S
= − = −
∂∂
7.47’
Pentru procesele ireversibile izobar-adiabatice, S = const., p = const., (sistem izolat adiabatic care achimbă cu mediul lucru mecanic la presiune constantă), folosind ecuaţia (7.45) putem scrie: dH TdS Vdp≤ + şi rezultă criteriul de echilibru: dH ≤ 0 sau:
H H TdS Vdpf ii
f
− < −∫ ( ) adică H Hf i− < 0 7.48
La echilibru: H = Hmin, dH = 0 d2H > 0 Deci, funcţia H care poartă numele de entalpie atinge un minim în starea de echilibru în condiţiile specificate. Proprietăţile termodinamice se pot determina din ecuaţiile (7.47) . ţinând seama că entalpia este o funcţie de stare din condiţia pentru diferenţială totală
exactă, pS
HSp
H∂∂
∂=
∂∂∂ 22
rezultă relaţia lui Maxwell:
;
∂∂
∂∂
=
∂∂
∂∂
pH
SSH
p
pS SV
pT
∂∂
=
∂∂
7.49
Derivând ecuaţia termică în raport cu p şi ecuaţia calorică în raport cu S se obţin coeficientul caloric Cp şi respectiv coeficientul termic KS:
;2
2
SH
ST
S ∂∂
=
∂∂ ;1
p
p
ST
TC
∂∂
⋅=ν
;1
2
2
SH
TC p
∂∂
⋅=ν
7.50
;2
2
pH
pV
S ∂∂
=
∂∂ ;1
SS p
VV
K
∂∂
−= 2
21pH
VK S ∂
∂⋅−= 7.51
7.2.4. Sistem termodinamic în contact cu un termostat: F(V,T) şi G(p,T) • Sistem în interacţie cu un termostat, la volum constant
Considerând un proces infinitezimal prin care sistemul trece dintr-o stare iniţială într-o stare finală apropiată, scriem ecuaţia fundamentală a termodinamicii: TdS dU L≥ + δ sau TdS dU pdV≥ +
d U TS TdS d TS pdV( ) ( )− ≤ − − 7.52 Pentru procese reversibile:
d U TS TdS d TS pdV( ) ( )− = − − 7.52’ Diferenţa (U-TS) se notează cu F şi poartă numele de energie liberă sau potenţial Helmholtz.
15
Deci, ecuaţia diferenţială a energiei libere F(V,T) este, din ecuaţia (7.52’): dF SdT pdV= − − 7.53
şi cum dF FT
dT FV
dVV T
=
+
∂∂
∂∂
7.54
Comparând ecuaţiile 7.53 şi 7.54 rezultă ecuaţia termică de stare şi exprimarea entropiei:
p FV T
= −
∂∂
şi S FT V
= −
∂∂
7.55
p şi S sunt parametri complementari corespunzători parametrilor V şi respectiv T. Ecuaţia calorică de stare sau ecuaţia Helmholtz se obţine imediat din definiţia energiei libere şi a entropiei:
U F TS= + şi deci U F 7.56 T FT V
= −
∂∂
Pentru procesele ireversibile izoterm-izocore, T = const., V = const. ecuaţia fundamentală se scrie:
dF SdT pdV≤ − − 7.57 De unde, criteriul de echilibru va fi:
dF ≤ 0 sau d U TS( )− < 0, adică dF < 0 sau F Ff i− < 0 7.58
prin urmare la echilibru termodinamic F îşi atinge minimul: F = Fmin, dF = 0 d2F> 0, 7.59
Deci, pentru un proces natural în condiţiile specificate, energia liberă a sistemului trebuie sa fie minimă în starea de echilibru. Proprietăţile termodinamice se pot determina din ecuaţiile (7.55) . Ţinând seama că energia liberă este o funcţie de stare, din condiţia pentru diferenţială
totală exactă, ∂∂ ∂
∂∂ ∂
2 2FV T
FT V
= rezultă relaţia lui Maxwell:
VT Tp
VS
VF
TTF
V
=
⇒
=
∂∂
∂∂
∂∂
∂∂
∂∂
∂∂
7.60
Din derivatele de ordinul al doilea ale funcţiei caracteristice energie liberă, care este şi potenţial termodinamic, se calculează căldura molară la volum constant CV şi coeficientul de compresibilitate izotermă, KT:
2
2
2
2
;TFTC
TSTC
TF
TS
VV
VV ∂
∂ν∂
∂ν∂
∂∂∂
−=
=⇒=
− 7.61
2
22
2 11 deci 1
TFV
KpV
VK
TF
Vp
TT
TT
∂∂∂
∂∂∂
∂−=
−=⇒=
∂
−
7.62
• Sistem în contact cu un termostat şi la presiune constantă. Sistemul termodinamic simplu are doi parametrii independenţi: presiunea şi temperatura. Pornind de la ecuaţia fundamentală se scrie:
d U pV TS SdT Vdp( )+ − ≤ − + 7.63
16
Marimea (U+pV-TS) poartă numele de entalpie liberă şi se notează cu G. G este potenţial termodinamic. Ca urmare, ecuaţia diferenţială a entalpiei libere va fi:
d U pV TS SdT Vdp( )+ − ≤ − + 7.64 şi ţinând seama că G(p,T) are diferenţială totală exactă:
dppGdT
TGdG
Tp
∂∂
+
∂∂
= 7.65
se pot obţine expresiile parametrilor complementari S şi V:
Tp pGV
TGS
∂∂
=
∂∂
−= si 7.66
A doua relaţie este ecuaţia termică de stare iar ecuaţia calorică se deduce imediat din expresia entalpiei libere şi poartă numele de ecuaţia Gibbs:
Tp pGp
TGTGUpVTSGU
∂∂
−
∂∂
−=−+= adica , 7.67
Pentru procesele ireversibile izoterm-izobare,T = const., p = const., ecuaţia fundamentală este:
dG SdT Vdp≤ − + 7.68 încât criteriul de echilibru va fi:
dG ≤ 0 7.69 sau,folosind ecuaţia pentru procese ireversibile se obţine: TdG < 0 sau Gf - Gi < 0 La echilibru, entalpia liberă îşi atinge minimul:
G = Gmin, dG = 0 d2G> 0 7.70 Echilibrul în starea considerată este deci caracterizat prin minimul entalpiei libere. Proprietăţile termodinamice se pot determina din ecuaţiile (7.66) . Ţinând seama că entalpia liberă este o funcţie de stare, din condiţia pentru diferenţială
totală exactă, ∂∂ ∂
∂∂ ∂
2 2Gp T
GT p
= rezultă relaţia lui Maxwell:
pT TV
pS
∂∂
=
∂∂
7.71
Din derivatele de ordinul al doilea ale funcţiei caracteristice entalpia liberă care este şi potential termodinamic, se calculează căldura molară la presiune constantă Cp şi coeficientul de compresibilitate izotermă, KT:
2
2
2
2
TGTC
TSTC
TG
TS
pp
pp ∂
∂ν∂
∂ν∂
∂∂∂
−=⇒
=⇒=
− 7.72
2
2
2
2 1 1 ;pG
VK
pV
VK
pG
pV
TT
TT ∂
∂∂∂
∂∂
∂∂
−=⇒
−==
7.73
7.2.5. Concluzii (Observaţii)
17
• Din punct de vedere practic, condiţiile impuse sistemului în cazul U(S,V) sunt dificil de regăsit în practică, în timp ce în cazul F(V,T) sunt cele mai frecvente; condiţiile impuse sistemului pentru H (p,S) sunt asigurate în reacţiile chimice în care nu se produce căldură şi nu au loc schimbări de fază; condiţiile impuse sistemului pentru G(p,T) sunt cele mai bune pentru studiul transformărilor de fază. • Principalele proprietăţi ale funcţiilor U(S,V), H(S,p), F(V,T) şi G(p,T): - sunt funcţii univoce de stare cu proprietatea de aditivitate (vom demonstra mai târziu); - derivatele lor de ordinul întâi în raport cu parametrii independenţi exprimă parametri complementari; - proprietatea fundamentală a acestor funcţii constă în aceea că la echilibru termodinamic aceste funcţii sunt minime (de aceea se numesc potentiale termodinamice); - descreşterea funcţiei în procesele compatibile cu condiţiile în care se află sistemul este măsurată de lucru mecanic efectuat de sistem; - În cazul în care, în afară de lucrul mecanic de destindere presupus anterior sistemul mai poate efectua un lucru mecanic de altă natură (în câmp electric, gravitaţional, etc.), atunci: δL = -pdV + δL’ astfel încât criteriile de echilibru se scriu: dP L+ ≤δ ' 0 unde P reprezintă unul dintre potenţialele U, H, F, G. - se pot deduce unele din altele; - conţin informaţia maximă despre sistemul termodinamic. Funcţiile Massieu şi potentialele termodinamice poartă numele de funcţii caracteristice.
Ca un corolar la concluziile precedente, • dacă un sistem complet izolat poate să existe în mai multe stări de echilibru, S
trebuie să fie aceeaşi în toate aceste stări ; • dacă un sistem la volum constant şi în contact cu un rezervor de căldură poate să existe în mai multe stări de echilibru stabil, F trebuie să fie aceeaşi în toate aceste stări; • dacă un sistem în contact cu un rezervor de căldură şi în mediu la presiune constantă poate să existe în mai multe stări de echilibru stabil,G trebuie să fie acelaşi în toate aceste stări. Se asociază aşadar, valori S, F, G sistemului numai pentru stările de echilibru stabil. Observaţie: Dacă noi putem construi funcţiile F, G, H, U pentru un sistem dat în condiţii specificate, atunci avem toate informaţiile termodinamice pentru sistem (ecuaţiile de stare). De exemplu, presupunem că un sistem închis într-o incintă adiabatică este format din două subsisteme cu temperaturi diferite, separate printr-un perete adiabatic. fiecare dintre subsisteme va tinde către o stare de echilibru stabil dar la temperaturi diferite. Peretele adiabatic care le separă constituie o constrângere care previne egalizarea temperaturilor. Un alt exemplu: presupunem că sistemul în contact cu un rezervor la temperatura T este de asemenea împărţit în doua subsisteme, fiecare conţinând (de exemplu) gaz la presiuni diferite. Ambele gaze (subsisteme) sunt în stare de echilibru
18
iar pistonul care le separă constituie o constrângere care nu permite egalizarea presiunilor. Al treilea exemplu presupune că de o parte şi de alta a peretelui despărţitor din cazul precedent există două gaze diferite, ambele la aceeaşi presiune. Dacă s-ar înlătura peretele, gazele ar difuza pâna când rezultă un amestec omogen. Peretele a constituit deci, o constrângere care a împiedicat omogenizarea. Dacă în primul exemplu este înlocuit peretele adiabatic, dacă se deplasează pistonul din al doilea exemplu sau se înlătură peretele despărţitor în al treilea caz, stările initiale au fost stări de echilibru, dar sistemul evoluează spontan către o noua stare de echilibru stabil, prin înlăturarea constrângerilor. În timpul procesului, când temperatura, presiunea sau compoziţia amestecului nu sunt uniforme, sistemul va fi în stare de neechilibru şi ca urmare unei asemenea stări nu putem asocia valori entropiei (S), energiei libere (F), şi entalpiei libere (G). Comparând stările de echilibru finală şi iniţială putem aplica concluziile deduse din criteriile de echilibru. Astfel, în primul exemplu, entropia finală va fi mai mare decât cea iniţială; în al doilea exemplu, dacă volumul sistemului este constant, valoarea finală a lui F va fi mai mică decât cea initială şi de asemenea în cel de-al treilea exemplu, G va atinge valoarea minimă. Se demonstrează că modalitatea de trecere de la un sistem de variabile naturale la altul (transformare care în matematică poartă numele de transformare Legendre) este singura care poate conserva toată informaţia termodinamică. Potenţialele termodinamice pot fi calculate pe cale termodinamică numai pentru două sisteme:gazul ideal şi radiaţia de echilibru, deoarece pentru acestea se cunosc ecuaţiile termică şi calorică de stare. Pentru toate celelalte sisteme potenţialele se află din experienţă sau cu ajutorul fizicii statistice. Pentru gaze funcţiile termodinamice se calculează cel mai des cu ajutorul fizicii statistice în timp ce pentru lichide şi solide ele se determină din experienţele de calorimetrie în care se determină căldurile specifice. De aici se vede legătura dintre termodinamică şi fizica statistică: ambele au acelaşi obiect de studiu, dar cercetarea în sine pleacă de la poziţii iniţiale deosebite. Pentru un studiu complet al proprietăţilor termodinamice trebuie folosite simultan atât termodinamica cât şi fizica statistică. 7.3. Proprietăţile funcţiilor caracteristice
19
În cazul general, sistemele termodinamice nu sunt simple, şi schimbă cu exteriorul atât energie cât şi substanţă (sisteme deschise). Astfel, este necesar un studiu privind dependenţa de masă a funcţiilor termodinamice caracteristice.
7.3.1. Dependenţa de masă a funcţiilor termodinamice caracteristice Vom studia pentru început stările de echilibru ale sistemelor omogene,
compuse dintr-o substanţă pură din punct de vedere chimic într-una din stările de agregare gazoasă, lichidă sau solidă; în ultimul caz vom presupune că singura solicitare mecanică constă într-o presiune uniform repartizată pe suprafaţa corpului. Vom neglija de asemenea, efectele datorită tensiunii superficiale. Ne vom referi pentru început la entropia sistemului de masă M (sau de număr de particule N) pentru care considerăm o dependenţă de forma:
S=S(U,V,M) 7.74 Mărimile S,U,V,M sunt mărimi extensive, fiind dependente de fracţionarea sau multiplicarea sistemului. Asta înseamnă că dacă fracţionăm sistemul în n părţi egale, identice din punct de vedere fizic, fiecare parte va avea volumul, energia, masa egale cu: V/ n, U/ n, M/ n, S/ n Dacă alipim m fracţiuni astfel încât să realizăm un nou sistem, acesta va avea V, U, M, S egale cu: mV/ n, mU/ n, mM/ n, mS/ n Rezultă deci că:
(S mn
V mn
U mn
M mn
S mn
S U V M, , , ,
= = )
)
7.75
sau notând m/ n = k S kU kV kM kS U V M( , , ) ( , ,= 7.76
Presupunând funcţia S continuă, relaţia (7.76) se generalizează de la valori pozitive şi raţionale ale lui k la valori pozitive arbitrare. Rezultă că funcţia S este omogenă de gradul I în raport cu variabilele de care depinde. Prin derivarea expresiei (7.76) în raport cu variabilele
)
U, V, M se obţine o relaţie de tipul:
S kU kV kM S U V Mα α' '( , , ) ( , ,= 7.77
unde α = U V M, ,
Exemplu: ;),,(),,(V
MVUSkV
kMkVkUSk∂
∂=
∂∂
UMVUSk
UkMkVkUSk
∂∂
=∂
∂ ),,(),,(
Rezultă că mărimile sunt funcţii omogene de grad zero, deci sunt intensive, adică nu depind de fracţionarea sau multiplicarea sistemului. Ţinând seama de forma diferenţială a entropiei în care considerăm şi masa ca variabilă :
Sα'
dS SU
dU SV
dV SN
dNV N U N U V
=
+
+
∂∂
∂∂
∂∂, , ,
7.78
şi de ecuaţiile:
1T
SU V
=
∂∂
şi pT
SV T
=
∂∂
7.79
rezultă că mărimile T şi p fiind determinate de sunt mărimi intensive. Sα'
20
Prin analogie cu ecuaţia (7.79) se defineşte o nouă mărime intensivă µ, denumită potenţial chimic:
TMS
VU
µ∂∂
−=
,
7.80
prin derivarea ambilor termeni din ecuaţia (7.76) în raport cu k şi luând k=1 se obţin ecuaţiile Euler:
MkM
kMkVkUS
VkV
kMkVkUSUkU
kMkVkUSMVUS
kVkU
kMkUkMkV
⋅
+⋅
+⋅
=
,
,,
)(),,(
)(),,(
)(),,(),,(
∂∂
∂∂
∂∂
7.81
şi pentru k=1:
S U V M U SU
V SV
M SMV M U M
( , , ), ,
= ⋅
+ ⋅
+ ⋅
∂∂
∂∂
∂∂
7.82
sau ţinând seama de relaţiile (7.79) şi (7.80):
S U V M UT
pVT
MT
( , , ) = + −µ 7.83
de unde se pot obţine ecuaţiile Euler pentru celelalte funcţii caracteristice:
U TS pV MH U pV TS MF U TS pV MG H TS M
= − += + = += − = − += − =
µµ
µµ
7.84
),( TpgM
TSpVU=
−+=µ 7.85
adică potenţialul chimic reprezintă entalpia liberă a unităţii de masă pentru starea dată. În general, din ecuaţiile de definiţie rezultă succesiv diferenţialele funcţiilor caracteristice:
T
dMpdVdUdS µ−+= 7.86
dMpdVTdSdU µ+−= 7.87 În membrul al doilea al relatiei (7.87) termenii au o semnificaţie simplă: -pdV=δLrev şi TdS = δQrev; unii autori interpretează termenul µdM ca o forma de lucru mecanic, în care M este coordonată generalizată iar µ forţa generalizată. Atunci, lucrul mecanic “total” va fi: (δLrev)total = -pdV+µdM şi ca urmare ecuaţia (7.87) exprimă principiul I.
dU = δLrev)total 7.87’ δQrev - (Deci putem spune că, dacă generalizăm notiunea de sistem termodinamic, atunci, pe lângă interacţiunile date de schimbul de lucru mecanic şi de schimbul de căldură în procesele considerate apare o nouă interacţie datorită schimbului de substanţă. În procesele cu schimb de masă este esenţial să se ţină seama de acest termen în aplicarea principiului I şi II.
dG SdT Vdp dM= − + + µ 7.90 Potenţialul chimic poate fi determinat, utilizându-se expresiile echivalente:
TpVTpSSV MMTpG
MMVTF
MMpSH
MMVSU
,,,,
),,(),,(),,(),,(
∂∂
=
∂∂
=
∂∂
=
∂∂
=µ
Întrucât entalpia liberă este o mărime intensivă în raport cu p, T şi extensivă în raport cu M se poate scrie relaţia:
G(p,T, M) = Mg(p,T) 7.92 unde g(p,T) este entalpia liberă a unităţii de masă. Prin urmare, considerându-se potenţialul chimic, se pot calcula variaţiile potenţialelor termodinamice funcţie de cantitatea de substanţă a sistemului termodinamic. Subliniem ca definiţiile anterioare ale proprietăţilor unei substanţe sunt valabile numai pentru stările de echilibru şi corespunzător acestor definiţii putem vorbi de S, G, H etc.
)
7.3.2. Proprietatea de aditivitate a funcţiilor caracteristice (potenţialelor termodinamice) În general un sistem termodinamic nu este simplu, având părţi constituiente neomogene. Se presupune întotdeauna că sistemul dat poate fi reprezentat ca un ansamblu termodinamic a unor sisteme omogene. Fie n sisteme simple în care poate fi descompus un sistem dat. Energia totală este întotdeauna suma energiilor componentelor. Deci,
dU dU kk
n
==
∑1
7.93
Aplicând pentru fiecare parte a sistemului expresia (7.76), se obţine:
dU T dS p dV dmk kk
n
k k k k= − +=
∑ (1
µ 7.94
Dar dU TdS pdV dM= − + µ unde T, p şi µ sunt parametrii sistemului compus. Rezulta de aici o ecuaţie generală de echilibru care poate fi folosită în toate situaţiile:
( )T dS p dV dm TdS pdV dMk kk
n
k k k k=
∑ − + = − +1
µ µ 7.95
Aditivitatea potenţialelor nu poate fi valabilă decât în condiţii bine precizate. De exemplu, dacă putem scrie relaţiile următoare: T T pentruk = , orice k, V V Mk
kk
k
= =∑ m∑; , atunci avem:
− − = − − −∑ ∑∑T dS dS p p dV dmkk
k k kkk
( ) ( ) (µ µ k) 7.96
şi dacă diferenţialele sunt independente, rezultă pk=p şi µk= µ oricare ar fi k şi de asemenea , de unde dS dSk
k
= ∑ S Skk
= ∑
Aşadar, echilibrul mecanic, chimic şi aditivitatea entropiei sunt condiţionate de uniformitatea temperaturii, de conservarea masei şi volumului. Într-o asemenea situaţie avem că:
dF S dT pdV dmk k k= − − k+ µ 7.97 şi deci
22
dF SdT pdV dM dFkk
∑ = − + =µ
de unde F k
k
= F∑ 7.97’
La fel se poate scrie pentru celelalte funcţii cracteristice: dH TdS V dp dmk k k= k+ + µ 7.98
dH TdS Vdp dM dHkk
∑ = + + =µ
de unde H Hk
k
= ∑ 7.98’
şi aşa mai departe. Dacă, de exemplu, sistemul va fi neomogen din punct de vedere termic, atunci
pentru că fenomenele de transport de cădură între părţile cu temperaturi
diferite sunt generatoare de entropie.
dS dSkk
≠ ∑
7.3.3. Ecuaţiile Gibbs-Duhem şi Duhem-Margule Pentru un sistem termodinamic compus din n sisteme simple energia totală este întotdeauna suma energiilor părţilor componente, astfel încât putem scrie:
dU dU kk
n
==
∑1
După cum am văzut în paragrafele precedente,
∑∑∑∑====
+−=+−=n
kkk
n
kkk
n
kk
n
kk dmdVpdSTdMpdVTdSdU
1111µµ
De obicei nu se poate preciza µ pentru exteriorul sistemului şi nici pk şi Vk
k
pentru fiecare subsistem. Atunci, putem privi sistemul compus ca datorat interacţiei mecanice şi calorice şi ca n interacţii chimice:
7.99 dU TdS pdV dmk
n
k= − +=
∑1
µ
În această ecuaţie, frecvent masa se înlocuieşte prin numărul de moli. Deci vom avea în loc de µk mărimi de acelaşi fel ~µ k , ~µ k = µk⋅(masa molară a componentului k) Dar, pentru a nu complica notaţiile vom scrie µk în loc de ~µ k , care reprezintă potenţialul chimic molar. Ca urmare, ecuaţia (7.99) se scrie:
dU TdS pdV dk
n
k= − +=
∑1
µ ν k 6.100
unde T şi V sunt temperatura şi volumul sistemului iar p presiunea pe care o exercita mediul asupra sistemului. Celelalte potenţiale termodinam
kνice vor avea formele diferenţiale:
dH S p TdS Vdp dk kk
( , , )ν µ= + + ∑
dF V T SdT pdV dkk
( , , )ν = − − + k kµ ν∑ 7.101
23
dG T p SdT Vdp dk kk
( , , )ν µ= − + + kν∑
Ţinând seama de ecuaţia Euler pentru V k( , , )νU S
U T , S pVk
n
k k= − +=
∑1
µ ν
pe care o diferenţiem, dU TdS SdT pdV Vdp d dk
n
k k k kk
n
= + − − + += =
∑ ∑1 1
µ ν ν µ
va rezulta ecuaţia Gibbs - Duhem:
SdT Vdp dk kk
n
− + ==
∑ν µ1
0 7.101’
sau sub forma:
− + ==
∑SdT Vdp dk kk
n
ν µ1
Relaţia Gibbs-Duhem este valabilă în toate condiţiile posibile ale sistemului. Dacă se scrie ecuaţia pentru presiune, temperatură şi masa totală, constante, se va obţine:
ν µk kk
n
d=
∑ =1
0 7.102
Dacă ţinem seama de ecuaţiile de stare atunci potenţialele chimice molare sunt funcţii de T, p şi . Se poate înlocui numărul de moli cu concentraţiile molare: ν k
ckk k
n
= =+ +
νν
νν ν ν1 2 .....
7.103
Deoarece masa totală este constantă, rezultă că:
ckk
n
=∑ =
1
1 7.104
Deci, din cele n concentraţii molare sunt independente numai n-1. Ca urmare avem: µ µk k nT p c c c= −( , , , ,.... )1 2 1 şi deci:
i
ijpT
n
i i
kn
ii
ijpTj
k
T
k
p
kk dc
cdc
cdp
pdT
Td
jjki ≠
−
=
−
=≠
∑
∂∂
=∑
∂∂
+
∂
∂+
=
,,,
1
1
1
1,,,,, νννν
µµµ∂∂µ
µ
Înlocuind d kµ în ecuaţia (7.102) vom avea:
ν
∂µ∂
νk
k
i T p j ii
n
k
n
icdc
j
=
≠=
−
=∑∑
, , ,1
1
1
0 7.106
Cum diferenţialele dci sunt independente, coeficienţii lor vor fi nuli:
1,.........2,1,0
,,,1
−==
∑
≠=
nic
cijpTi
kn
kk
jν∂∂µ
7.107
24
Acestea sunt relaţiile Duhem-Margule. Ele au o importanţă deosebită în chimia-fizică. De exemplu se poate demonstra că pentru un anumit tip de amestec, µk sunt funcţii numai de T, p şi ck. Deci:
∂µ∂
∂µ∂
δk
i
k
kkic c
= 7.108
iar din relaţiile precedente se obţine că pentru amestecurile ideale avem:
cck
k
nk
k T p=∑
=
1
0∂µ∂ ,
7.109
Relaţia (7.109) poate fi folosită pentru determinarea dependenţei potenţialului chimic de concentraţii.