

Anexa I
Lista cu denumirile comerciale, forma(ele) farmaceutică(e), concentraţia(iile), calea(căile) de administrare a(ale) medicamentului (elor),
solicitantul (ţii), deţinătorul(ii) autorizaţiei de punere pe piaţă în statele member
1
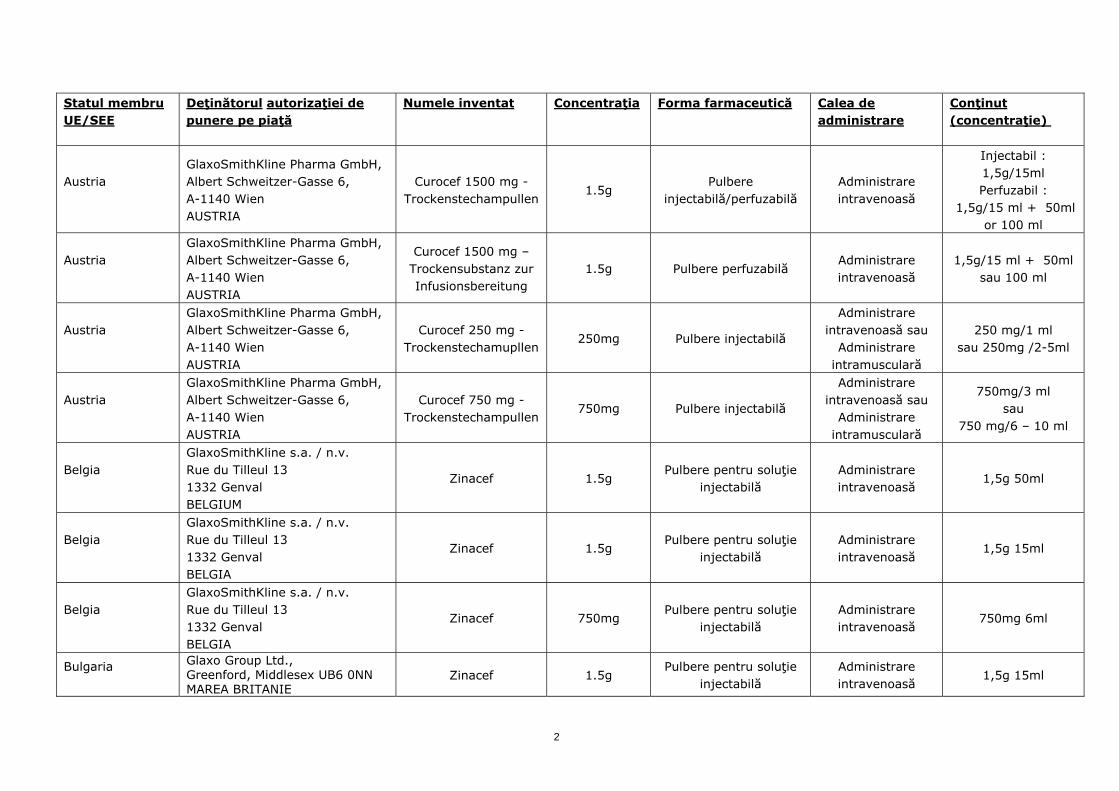
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
Austria
GlaxoSmithKline Pharma GmbH, Albert Schweitzer-Gasse 6, A-1140 Wien AUSTRIA
Curocef 1500 mg - Trockenstechampullen
1.5g Pulbere
injectabilă/perfuzabilă Administrare intravenoasă
Injectabil : 1,5g/15ml Perfuzabil :
1,5g/15 ml + 50ml or 100 ml
Austria
GlaxoSmithKline Pharma GmbH, Albert Schweitzer-Gasse 6, A-1140 Wien AUSTRIA
Curocef 1500 mg – Trockensubstanz zur Infusionsbereitung
1.5g Pulbere perfuzabilă Administrare intravenoasă
1,5g/15 ml + 50ml sau 100 ml
Austria
GlaxoSmithKline Pharma GmbH, Albert Schweitzer-Gasse 6, A-1140 Wien AUSTRIA
Curocef 250 mg - Trockenstechamupllen
250mg Pulbere injectabilă
Administrare intravenoasă sau
Administrare intramusculară
250 mg/1 ml sau 250mg /2-5ml
Austria
GlaxoSmithKline Pharma GmbH, Albert Schweitzer-Gasse 6, A-1140 Wien AUSTRIA
Curocef 750 mg - Trockenstechampullen
750mg Pulbere injectabilă
Administrare intravenoasă sau
Administrare intramusculară
750mg/3 ml sau
750 mg/6 – 10 ml
Belgia
GlaxoSmithKline s.a. / n.v. Rue du Tilleul 13 1332 Genval BELGIUM
Zinacef 1.5g Pulbere pentru soluţie
injectabilă Administrare intravenoasă
1,5g 50ml
Belgia
GlaxoSmithKline s.a. / n.v. Rue du Tilleul 13 1332 Genval BELGIA
Zinacef 1.5g Pulbere pentru soluţie
injectabilă Administrare intravenoasă
1,5g 15ml
Belgia
GlaxoSmithKline s.a. / n.v. Rue du Tilleul 13 1332 Genval BELGIA
Zinacef 750mg Pulbere pentru soluţie
injectabilă Administrare intravenoasă
750mg 6ml
Bulgaria
Glaxo Group Ltd., Greenford, Middlesex UB6 0NN MAREA BRITANIE
Zinacef 1.5g Pulbere pentru soluţie
injectabilă Administrare intravenoasă
1,5g 15ml
2
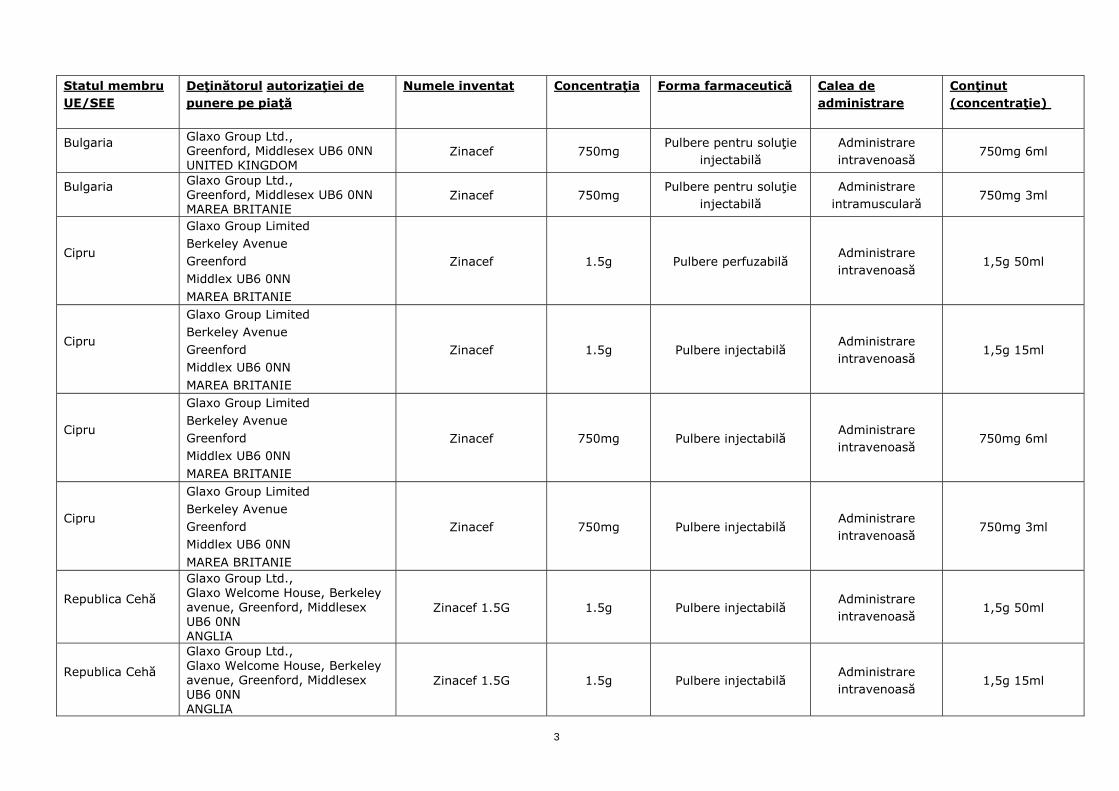
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
Bulgaria
Glaxo Group Ltd., Greenford, Middlesex UB6 0NN UNITED KINGDOM
Zinacef 750mg Pulbere pentru soluţie
injectabilă Administrare intravenoasă
750mg 6ml
Bulgaria
Glaxo Group Ltd., Greenford, Middlesex UB6 0NN MAREA BRITANIE
Zinacef 750mg Pulbere pentru soluţie
injectabilă Administrare
intramusculară 750mg 3ml
Cipru
Glaxo Group Limited Berkeley Avenue Greenford Middlex UB6 0NN MAREA BRITANIE
Zinacef 1.5g Pulbere perfuzabilă Administrare intravenoasă
1,5g 50ml
Cipru
Glaxo Group Limited Berkeley Avenue Greenford Middlex UB6 0NN MAREA BRITANIE
Zinacef 1.5g Pulbere injectabilă Administrare intravenoasă
1,5g 15ml
Cipru
Glaxo Group Limited Berkeley Avenue Greenford Middlex UB6 0NN MAREA BRITANIE
Zinacef 750mg Pulbere injectabilă Administrare intravenoasă
750mg 6ml
Cipru
Glaxo Group Limited Berkeley Avenue Greenford Middlex UB6 0NN MAREA BRITANIE
Zinacef 750mg Pulbere injectabilă Administrare intravenoasă
750mg 3ml
Republica Cehă
Glaxo Group Ltd., Glaxo Welcome House, Berkeley avenue, Greenford, Middlesex UB6 0NN ANGLIA
Zinacef 1.5G 1.5g Pulbere injectabilă Administrare intravenoasă
1,5g 50ml
Republica Cehă
Glaxo Group Ltd., Glaxo Welcome House, Berkeley avenue, Greenford, Middlesex UB6 0NN ANGLIA
Zinacef 1.5G 1.5g Pulbere injectabilă Administrare intravenoasă
1,5g 15ml
3
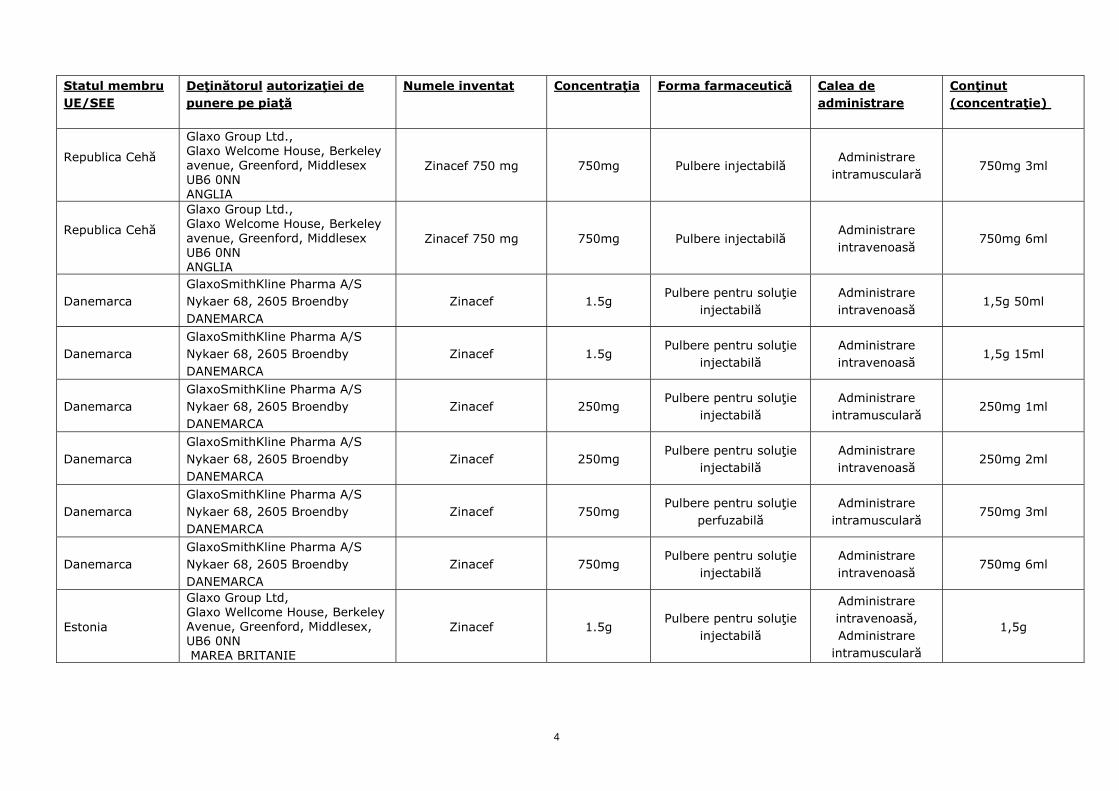
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
Republica Cehă
Glaxo Group Ltd., Glaxo Welcome House, Berkeley avenue, Greenford, Middlesex UB6 0NN ANGLIA
Zinacef 750 mg 750mg Pulbere injectabilă Administrare
intramusculară 750mg 3ml
Republica Cehă
Glaxo Group Ltd., Glaxo Welcome House, Berkeley avenue, Greenford, Middlesex UB6 0NN ANGLIA
Zinacef 750 mg 750mg Pulbere injectabilă Administrare intravenoasă
750mg 6ml
Danemarca GlaxoSmithKline Pharma A/S Nykaer 68, 2605 Broendby DANEMARCA
Zinacef 1.5g Pulbere pentru soluţie
injectabilă Administrare intravenoasă
1,5g 50ml
Danemarca GlaxoSmithKline Pharma A/S Nykaer 68, 2605 Broendby DANEMARCA
Zinacef 1.5g Pulbere pentru soluţie
injectabilă Administrare intravenoasă
1,5g 15ml
Danemarca GlaxoSmithKline Pharma A/S Nykaer 68, 2605 Broendby DANEMARCA
Zinacef 250mg Pulbere pentru soluţie
injectabilă Administrare
intramusculară 250mg 1ml
Danemarca GlaxoSmithKline Pharma A/S Nykaer 68, 2605 Broendby DANEMARCA
Zinacef 250mg Pulbere pentru soluţie
injectabilă Administrare intravenoasă
250mg 2ml
Danemarca GlaxoSmithKline Pharma A/S Nykaer 68, 2605 Broendby DANEMARCA
Zinacef 750mg Pulbere pentru soluţie
perfuzabilă Administrare
intramusculară 750mg 3ml
Danemarca GlaxoSmithKline Pharma A/S Nykaer 68, 2605 Broendby DANEMARCA
Zinacef 750mg Pulbere pentru soluţie
injectabilă Administrare intravenoasă
750mg 6ml
Estonia
Glaxo Group Ltd, Glaxo Wellcome House, Berkeley Avenue, Greenford, Middlesex, UB6 0NN MAREA BRITANIE
Zinacef 1.5g Pulbere pentru soluţie
injectabilă
Administrare intravenoasă, Administrare
intramusculară
1,5g
4
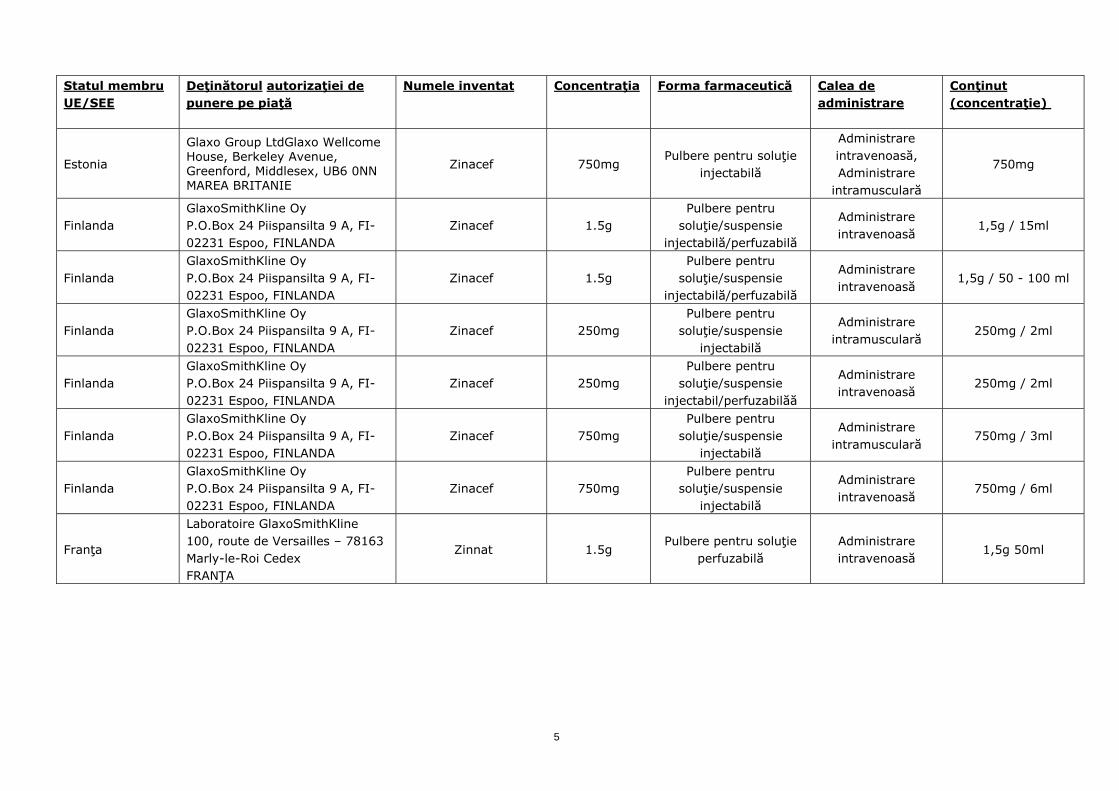
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
Estonia
Glaxo Group LtdGlaxo Wellcome House, Berkeley Avenue, Greenford, Middlesex, UB6 0NN MAREA BRITANIE
Zinacef 750mg Pulbere pentru soluţie
injectabilă
Administrare intravenoasă, Administrare
intramusculară
750mg
Finlanda GlaxoSmithKline Oy P.O.Box 24 Piispansilta 9 A, FI-02231 Espoo, FINLANDA
Zinacef 1.5g Pulbere pentru
soluţie/suspensie injectabilă/perfuzabilă
Administrare intravenoasă
1,5g / 15ml
Finlanda GlaxoSmithKline Oy P.O.Box 24 Piispansilta 9 A, FI-02231 Espoo, FINLANDA
Zinacef 1.5g Pulbere pentru
soluţie/suspensie injectabilă/perfuzabilă
Administrare intravenoasă
1,5g / 50 - 100 ml
Finlanda GlaxoSmithKline Oy P.O.Box 24 Piispansilta 9 A, FI-02231 Espoo, FINLANDA
Zinacef 250mg Pulbere pentru
soluţie/suspensie injectabilă
Administrare intramusculară
250mg / 2ml
Finlanda GlaxoSmithKline Oy P.O.Box 24 Piispansilta 9 A, FI-02231 Espoo, FINLANDA
Zinacef 250mg Pulbere pentru
soluţie/suspensie injectabil/perfuzabilăă
Administrare intravenoasă
250mg / 2ml
Finlanda GlaxoSmithKline Oy P.O.Box 24 Piispansilta 9 A, FI-02231 Espoo, FINLANDA
Zinacef 750mg Pulbere pentru
soluţie/suspensie injectabilă
Administrare intramusculară
750mg / 3ml
Finlanda GlaxoSmithKline Oy P.O.Box 24 Piispansilta 9 A, FI-02231 Espoo, FINLANDA
Zinacef 750mg Pulbere pentru
soluţie/suspensie injectabilă
Administrare intravenoasă
750mg / 6ml
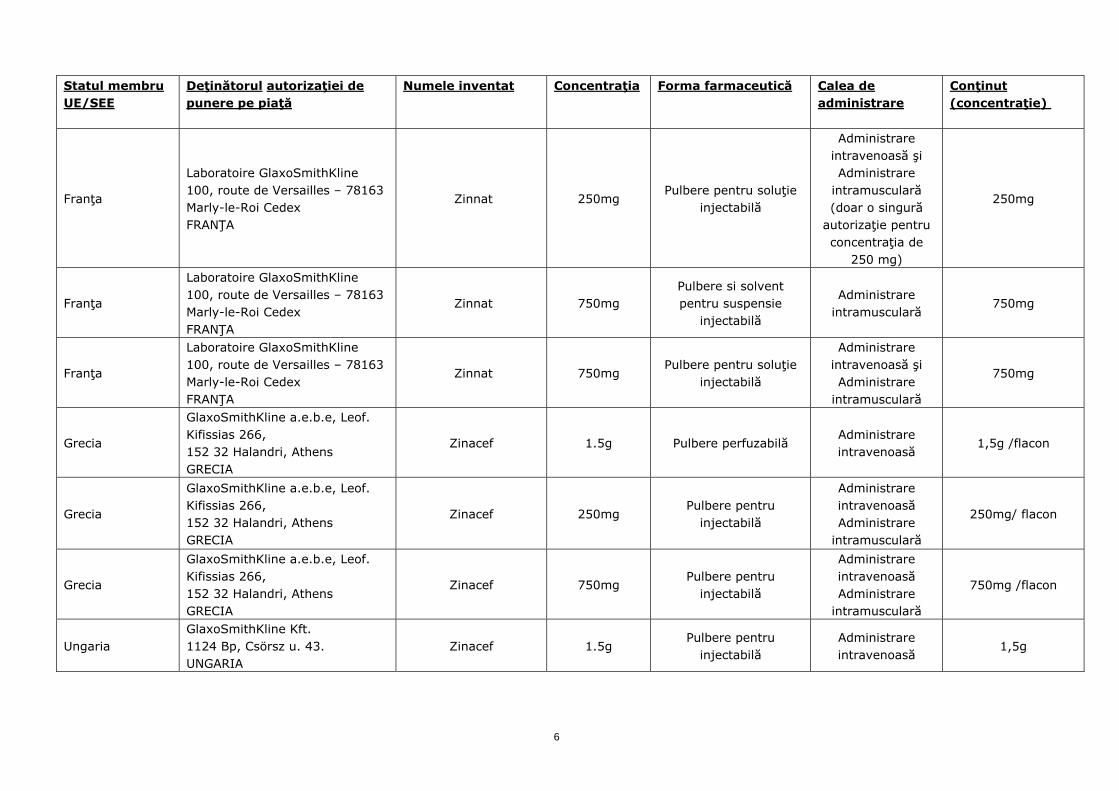
Franţa
Laboratoire GlaxoSmithKline 100, route de Versailles – 78163 Marly-le-Roi Cedex FRANŢA
Zinnat 1.5g Pulbere pentru soluţie
perfuzabilă Administrare intravenoasă
1,5g 50ml
5
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
Franţa
Laboratoire GlaxoSmithKline 100, route de Versailles – 78163 Marly-le-Roi Cedex FRANŢA
Zinnat 250mg Pulbere pentru soluţie
injectabilă
Administrare intravenoasă şi Administrare
intramusculară (doar o singură
autorizaţie pentru concentraţia de
250 mg)
250mg
Franţa
Laboratoire GlaxoSmithKline 100, route de Versailles – 78163 Marly-le-Roi Cedex FRANŢA
Zinnat 750mg Pulbere si solvent pentru suspensie
injectabilă
Administrare intramusculară
750mg
Franţa
Laboratoire GlaxoSmithKline 100, route de Versailles – 78163 Marly-le-Roi Cedex FRANŢA
Zinnat 750mg Pulbere pentru soluţie
injectabilă
Administrare intravenoasă şi Administrare
intramusculară
750mg
Grecia
GlaxoSmithKline a.e.b.e, Leof. Kifissias 266, 152 32 Halandri, Athens GRECIA
Zinacef 1.5g Pulbere perfuzabilă Administrare intravenoasă
1,5g /flacon
Grecia
GlaxoSmithKline a.e.b.e, Leof. Kifissias 266, 152 32 Halandri, Athens GRECIA
Zinacef 250mg Pulbere pentru
injectabilă
Administrare intravenoasă Administrare
intramusculară
250mg/ flacon
Grecia
GlaxoSmithKline a.e.b.e, Leof. Kifissias 266, 152 32 Halandri, Athens GRECIA
Zinacef 750mg Pulbere pentru
injectabilă
Administrare intravenoasă Administrare
intramusculară
750mg /flacon
Ungaria GlaxoSmithKline Kft. 1124 Bp, Csörsz u. 43. UNGARIA
Zinacef 1.5g Pulbere pentru
injectabilă Administrare intravenoasă
1,5g
6
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
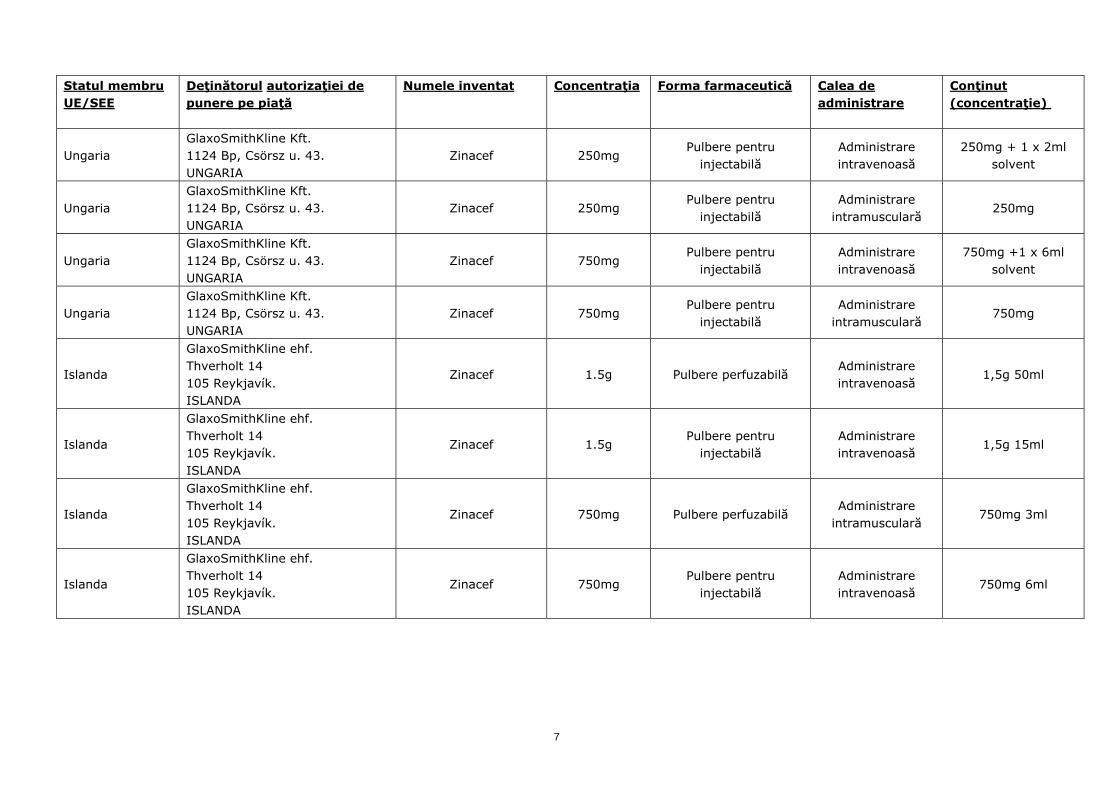
Ungaria GlaxoSmithKline Kft. 1124 Bp, Csörsz u. 43. UNGARIA
Zinacef 250mg Pulbere pentru
injectabilă Administrare intravenoasă
250mg + 1 x 2ml solvent
Ungaria GlaxoSmithKline Kft. 1124 Bp, Csörsz u. 43. UNGARIA
Zinacef 250mg Pulbere pentru
injectabilă Administrare
intramusculară 250mg
Ungaria GlaxoSmithKline Kft. 1124 Bp, Csörsz u. 43. UNGARIA
Zinacef 750mg Pulbere pentru
injectabilă Administrare intravenoasă
750mg +1 x 6ml solvent
Ungaria GlaxoSmithKline Kft. 1124 Bp, Csörsz u. 43. UNGARIA
Zinacef 750mg Pulbere pentru
injectabilă Administrare
intramusculară 750mg
Islanda
GlaxoSmithKline ehf. Thverholt 14 105 Reykjavík. ISLANDA
Zinacef 1.5g Pulbere perfuzabilă Administrare intravenoasă
1,5g 50ml
Islanda
GlaxoSmithKline ehf. Thverholt 14 105 Reykjavík. ISLANDA
Zinacef 1.5g Pulbere pentru
injectabilă Administrare intravenoasă
1,5g 15ml
Islanda
GlaxoSmithKline ehf. Thverholt 14 105 Reykjavík. ISLANDA
Zinacef 750mg Pulbere perfuzabilă Administrare
intramusculară 750mg 3ml
Islanda
GlaxoSmithKline ehf. Thverholt 14 105 Reykjavík. ISLANDA
Zinacef 750mg Pulbere pentru
injectabilă Administrare intravenoasă
750mg 6ml
7
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
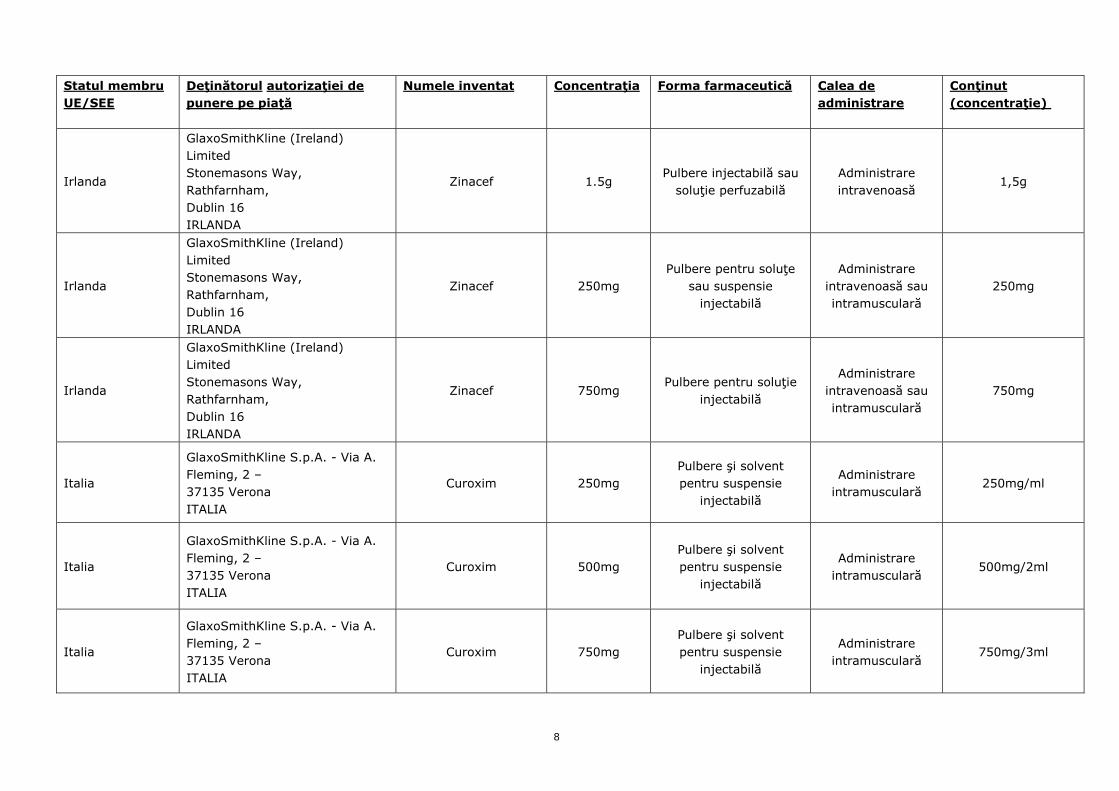
Irlanda
GlaxoSmithKline (Ireland) Limited Stonemasons Way, Rathfarnham, Dublin 16 IRLANDA
Zinacef 1.5g Pulbere injectabilă sau
soluţie perfuzabilă Administrare intravenoasă
1,5g
Irlanda
GlaxoSmithKline (Ireland) Limited Stonemasons Way, Rathfarnham, Dublin 16 IRLANDA
Zinacef 250mg Pulbere pentru soluţe
sau suspensie injectabilă
Administrare intravenoasă sau intramusculară
250mg
Irlanda
GlaxoSmithKline (Ireland) Limited Stonemasons Way, Rathfarnham, Dublin 16 IRLANDA
Zinacef 750mg Pulbere pentru soluţie
injectabilă
Administrare intravenoasă sau intramusculară
750mg
Italia
GlaxoSmithKline S.p.A. - Via A. Fleming, 2 – 37135 Verona ITALIA
Curoxim 250mg Pulbere şi solvent pentru suspensie
injectabilă
Administrare intramusculară
250mg/ml
Italia
GlaxoSmithKline S.p.A. - Via A. Fleming, 2 – 37135 Verona ITALIA
Curoxim 500mg Pulbere şi solvent pentru suspensie
injectabilă
Administrare intramusculară
500mg/2ml
Italia
GlaxoSmithKline S.p.A. - Via A. Fleming, 2 – 37135 Verona ITALIA
Curoxim 750mg Pulbere şi solvent pentru suspensie
injectabilă
Administrare intramusculară
750mg/3ml
8
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
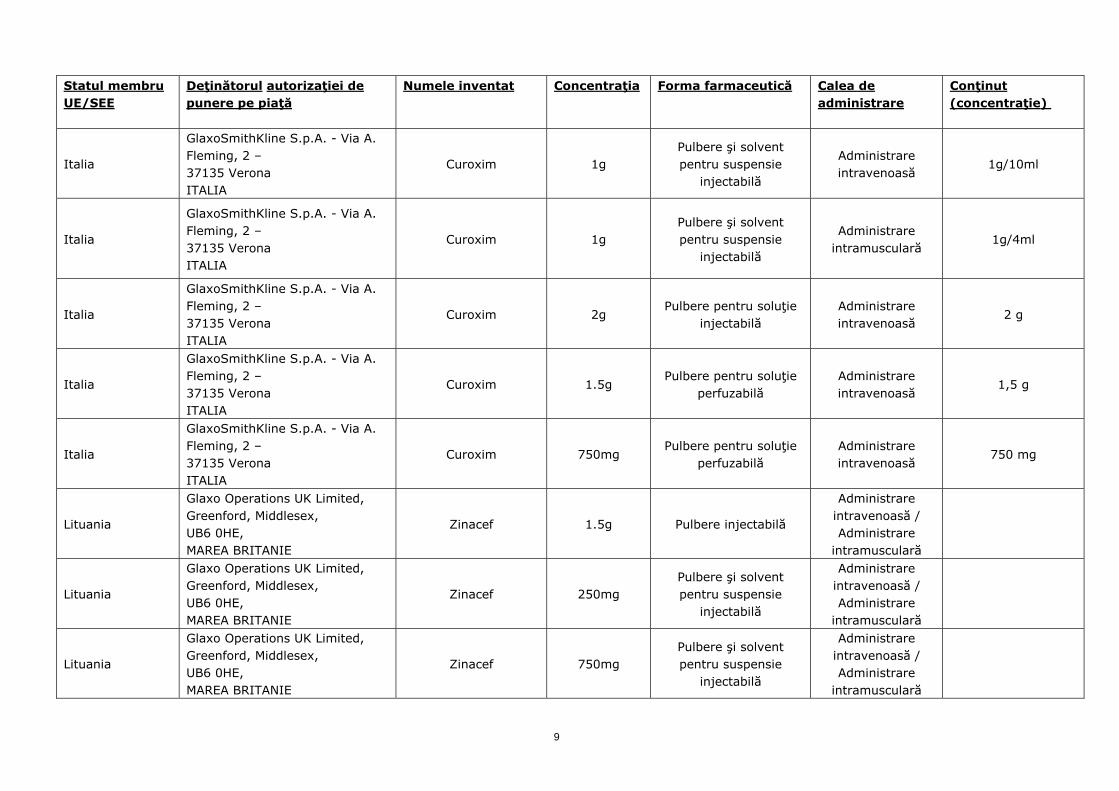
Italia
GlaxoSmithKline S.p.A. - Via A. Fleming, 2 – 37135 Verona ITALIA
Curoxim 1g Pulbere şi solvent pentru suspensie
injectabilă
Administrare intravenoasă
1g/10ml
Italia
GlaxoSmithKline S.p.A. - Via A. Fleming, 2 – 37135 Verona ITALIA
Curoxim 1g Pulbere şi solvent pentru suspensie
injectabilă
Administrare intramusculară
1g/4ml
Italia
GlaxoSmithKline S.p.A. - Via A. Fleming, 2 – 37135 Verona ITALIA
Curoxim 2g Pulbere pentru soluţie
injectabilă Administrare intravenoasă
2 g
Italia
GlaxoSmithKline S.p.A. - Via A. Fleming, 2 – 37135 Verona ITALIA
Curoxim 1.5g Pulbere pentru soluţie
perfuzabilă Administrare intravenoasă
1,5 g
Italia
GlaxoSmithKline S.p.A. - Via A. Fleming, 2 – 37135 Verona ITALIA
Curoxim 750mg Pulbere pentru soluţie
perfuzabilă Administrare intravenoasă
750 mg
Lituania
Glaxo Operations UK Limited, Greenford, Middlesex, UB6 0HE, MAREA BRITANIE
Zinacef 1.5g Pulbere injectabilă
Administrare intravenoasă / Administrare
intramusculară
Lituania
Glaxo Operations UK Limited, Greenford, Middlesex, UB6 0HE, MAREA BRITANIE
Zinacef 250mg Pulbere şi solvent pentru suspensie
injectabilă
Administrare intravenoasă / Administrare
intramusculară
Lituania
Glaxo Operations UK Limited, Greenford, Middlesex, UB6 0HE, MAREA BRITANIE
Zinacef 750mg Pulbere şi solvent pentru suspensie
injectabilă
Administrare intravenoasă / Administrare
intramusculară
9
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
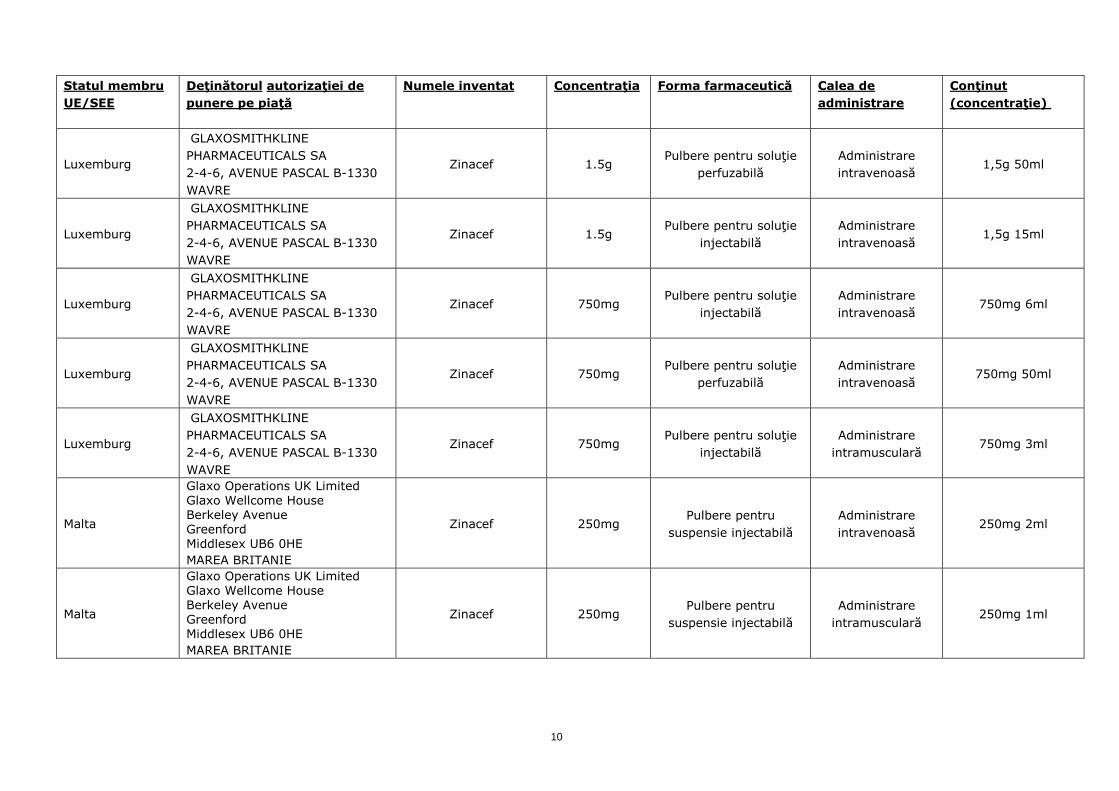
Luxemburg
GLAXOSMITHKLINE PHARMACEUTICALS SA 2-4-6, AVENUE PASCAL B-1330 WAVRE
Zinacef 1.5g Pulbere pentru soluţie
perfuzabilă Administrare intravenoasă
1,5g 50ml
Luxemburg
GLAXOSMITHKLINE PHARMACEUTICALS SA 2-4-6, AVENUE PASCAL B-1330 WAVRE
Zinacef 1.5g Pulbere pentru soluţie
injectabilă Administrare intravenoasă
1,5g 15ml
Luxemburg
GLAXOSMITHKLINE PHARMACEUTICALS SA 2-4-6, AVENUE PASCAL B-1330 WAVRE
Zinacef 750mg Pulbere pentru soluţie
injectabilă Administrare intravenoasă
750mg 6ml
Luxemburg
GLAXOSMITHKLINE PHARMACEUTICALS SA 2-4-6, AVENUE PASCAL B-1330 WAVRE
Zinacef 750mg Pulbere pentru soluţie
perfuzabilă Administrare intravenoasă
750mg 50ml
Luxemburg
GLAXOSMITHKLINE PHARMACEUTICALS SA 2-4-6, AVENUE PASCAL B-1330 WAVRE
Zinacef 750mg Pulbere pentru soluţie
injectabilă Administrare
intramusculară 750mg 3ml
Malta
Glaxo Operations UK Limited Glaxo Wellcome House Berkeley Avenue Greenford Middlesex UB6 0HE MAREA BRITANIE
Zinacef 250mg Pulbere pentru
suspensie injectabilă Administrare intravenoasă
250mg 2ml
Malta
Glaxo Operations UK Limited Glaxo Wellcome House Berkeley Avenue Greenford Middlesex UB6 0HE MAREA BRITANIE
Zinacef 250mg Pulbere pentru
suspensie injectabilă Administrare
intramusculară 250mg 1ml
10
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
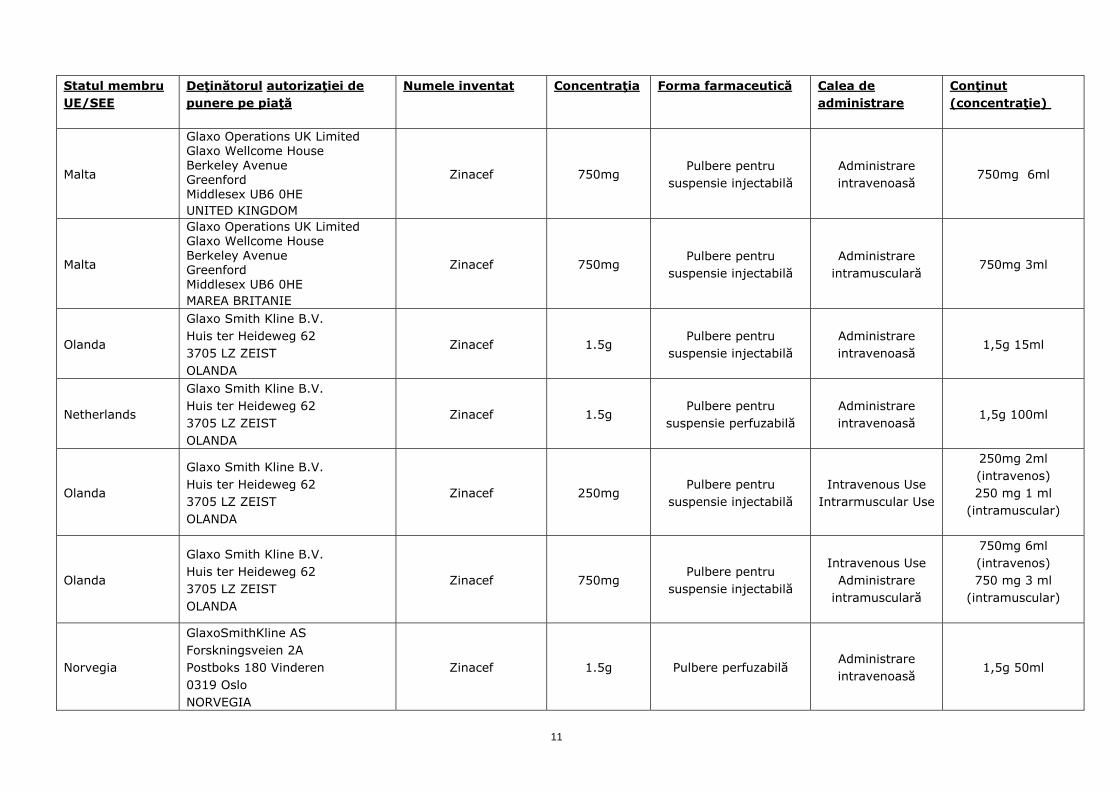
Malta
Glaxo Operations UK Limited Glaxo Wellcome House Berkeley Avenue Greenford Middlesex UB6 0HE UNITED KINGDOM
Zinacef 750mg Pulbere pentru
suspensie injectabilă Administrare intravenoasă
750mg 6ml
Malta
Glaxo Operations UK Limited Glaxo Wellcome House Berkeley Avenue Greenford Middlesex UB6 0HE MAREA BRITANIE
Zinacef 750mg Pulbere pentru
suspensie injectabilă Administrare
intramusculară 750mg 3ml
Olanda
Glaxo Smith Kline B.V. Huis ter Heideweg 62 3705 LZ ZEIST OLANDA
Zinacef 1.5g Pulbere pentru
suspensie injectabilă Administrare intravenoasă
1,5g 15ml
Netherlands
Glaxo Smith Kline B.V. Huis ter Heideweg 62 3705 LZ ZEIST OLANDA
Zinacef 1.5g Pulbere pentru
suspensie perfuzabilă Administrare intravenoasă
1,5g 100ml
Olanda
Glaxo Smith Kline B.V. Huis ter Heideweg 62 3705 LZ ZEIST OLANDA
Zinacef 250mg Pulbere pentru
suspensie injectabilă Intravenous Use
Intrarmuscular Use
250mg 2ml (intravenos) 250 mg 1 ml
(intramuscular)
Olanda
Glaxo Smith Kline B.V. Huis ter Heideweg 62 3705 LZ ZEIST OLANDA
Zinacef 750mg Pulbere pentru
suspensie injectabilă
Intravenous Use Administrare
intramusculară
750mg 6ml (intravenos) 750 mg 3 ml
(intramuscular)
Norvegia
GlaxoSmithKline AS Forskningsveien 2A Postboks 180 Vinderen 0319 Oslo NORVEGIA
Zinacef 1.5g Pulbere perfuzabilă Administrare intravenoasă
1,5g 50ml
11
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
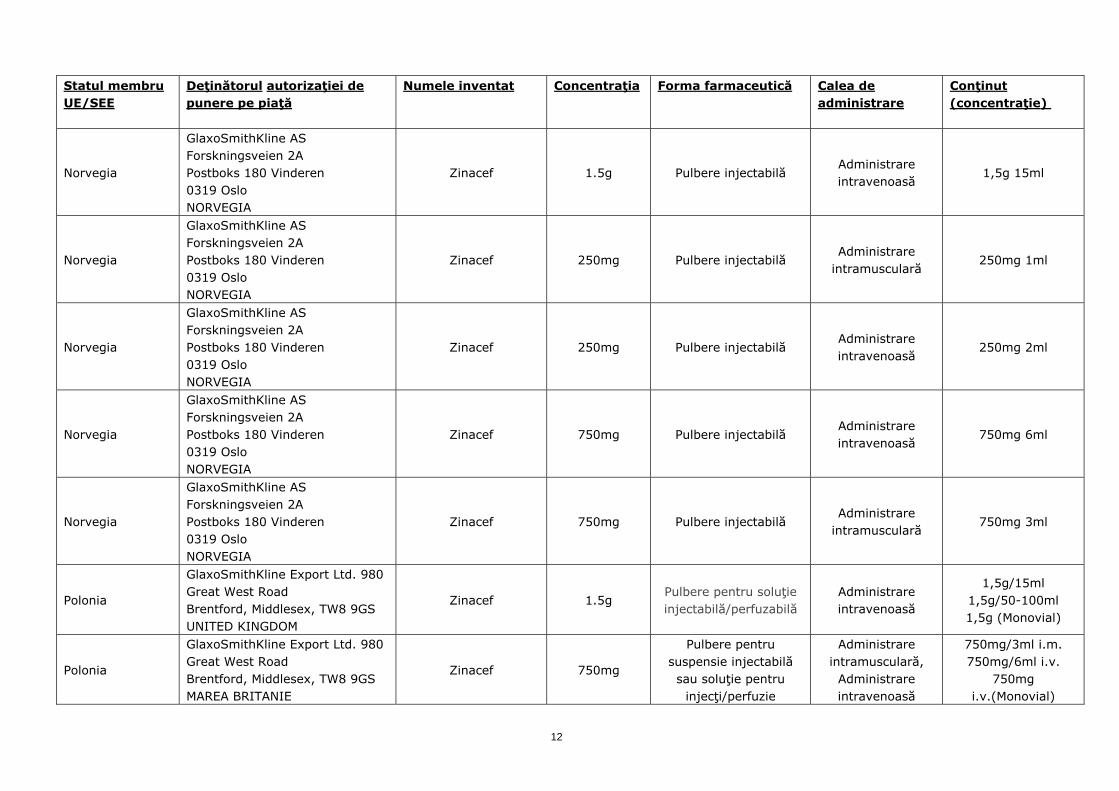
Norvegia
GlaxoSmithKline AS Forskningsveien 2A Postboks 180 Vinderen 0319 Oslo NORVEGIA
Zinacef 1.5g Pulbere injectabilă Administrare intravenoasă
1,5g 15ml
Norvegia
GlaxoSmithKline AS Forskningsveien 2A Postboks 180 Vinderen 0319 Oslo NORVEGIA
Zinacef 250mg Pulbere injectabilă Administrare
intramusculară 250mg 1ml
Norvegia
GlaxoSmithKline AS Forskningsveien 2A Postboks 180 Vinderen 0319 Oslo NORVEGIA
Zinacef 250mg Pulbere injectabilă Administrare intravenoasă
250mg 2ml
Norvegia
GlaxoSmithKline AS Forskningsveien 2A Postboks 180 Vinderen 0319 Oslo NORVEGIA
Zinacef 750mg Pulbere injectabilă Administrare intravenoasă
750mg 6ml
Norvegia
GlaxoSmithKline AS Forskningsveien 2A Postboks 180 Vinderen 0319 Oslo NORVEGIA
Zinacef 750mg Pulbere injectabilă Administrare
intramusculară 750mg 3ml
Polonia
GlaxoSmithKline Export Ltd. 980 Great West Road Brentford, Middlesex, TW8 9GS UNITED KINGDOM
Zinacef 1.5g Pulbere pentru soluţie injectabilă/perfuzabilă
Administrare intravenoasă
1,5g/15ml 1,5g/50-100ml 1,5g (Monovial)
Polonia
GlaxoSmithKline Export Ltd. 980 Great West Road Brentford, Middlesex, TW8 9GS MAREA BRITANIE
Zinacef 750mg
Pulbere pentru suspensie injectabilă sau soluţie pentru
injecţi/perfuzie
Administrare intramusculară,
Administrare intravenoasă
750mg/3ml i.m. 750mg/6ml i.v.
750mg i.v.(Monovial)
12
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
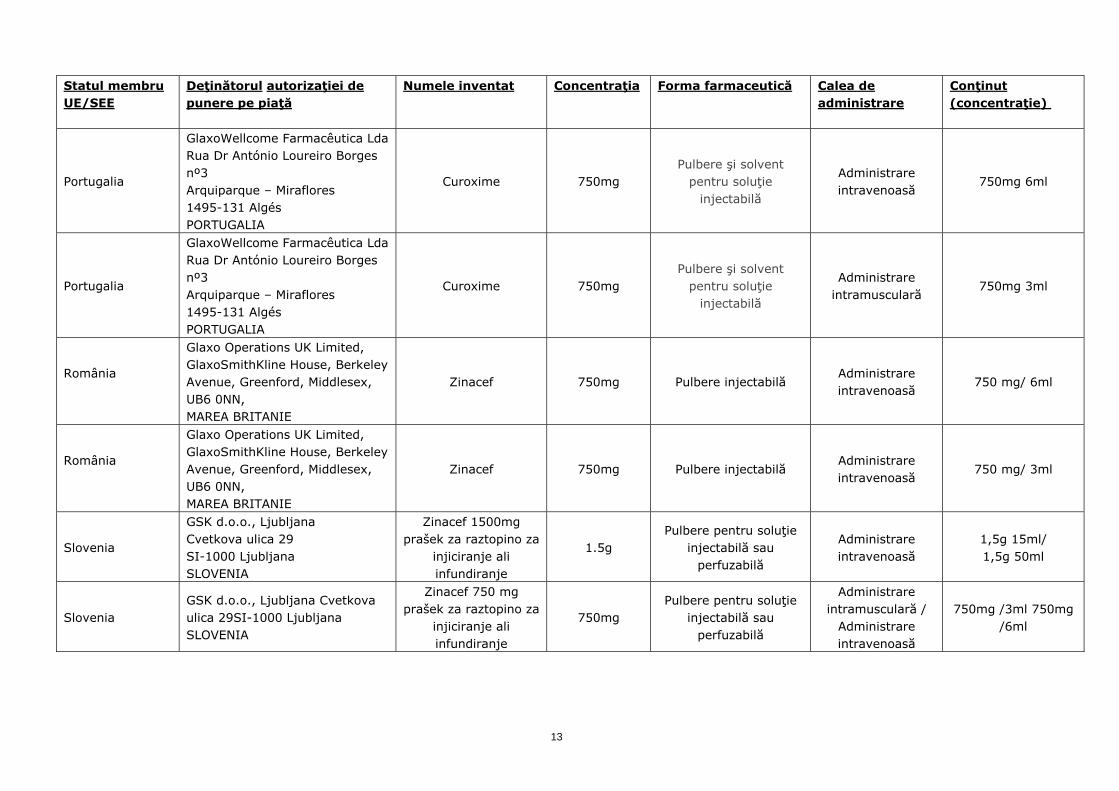
Portugalia
GlaxoWellcome Farmacêutica Lda Rua Dr António Loureiro Borges nº3 Arquiparque – Miraflores 1495-131 Algés PORTUGALIA
Curoxime 750mg Pulbere şi solvent
pentru soluţie injectabilă
Administrare intravenoasă
750mg 6ml
Portugalia
GlaxoWellcome Farmacêutica Lda Rua Dr António Loureiro Borges nº3 Arquiparque – Miraflores 1495-131 Algés PORTUGALIA
Curoxime 750mg Pulbere şi solvent
pentru soluţie injectabilă
Administrare intramusculară
750mg 3ml
România
Glaxo Operations UK Limited, GlaxoSmithKline House, Berkeley Avenue, Greenford, Middlesex, UB6 0NN, MAREA BRITANIE
Zinacef 750mg Pulbere injectabilă Administrare intravenoasă
750 mg/ 6ml
România
Glaxo Operations UK Limited, GlaxoSmithKline House, Berkeley Avenue, Greenford, Middlesex, UB6 0NN, MAREA BRITANIE
Zinacef 750mg Pulbere injectabilă Administrare intravenoasă
750 mg/ 3ml
Slovenia
GSK d.o.o., Ljubljana Cvetkova ulica 29 SI-1000 Ljubljana SLOVENIA
Zinacef 1500mg prašek za raztopino za
injiciranje ali infundiranje
1.5g Pulbere pentru soluţie
injectabilă sau perfuzabilă
Administrare intravenoasă
1,5g 15ml/ 1,5g 50ml
Slovenia GSK d.o.o., Ljubljana Cvetkova ulica 29SI-1000 Ljubljana SLOVENIA
Zinacef 750 mg prašek za raztopino za
injiciranje ali infundiranje
750mg Pulbere pentru soluţie
injectabilă sau perfuzabilă
Administrare intramusculară /
Administrare intravenoasă
750mg /3ml 750mg /6ml
13
14
Statul membru UE/SEE
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat
Concentraţia Forma farmaceutică Calea de administrare
Conţinut (concentraţie)
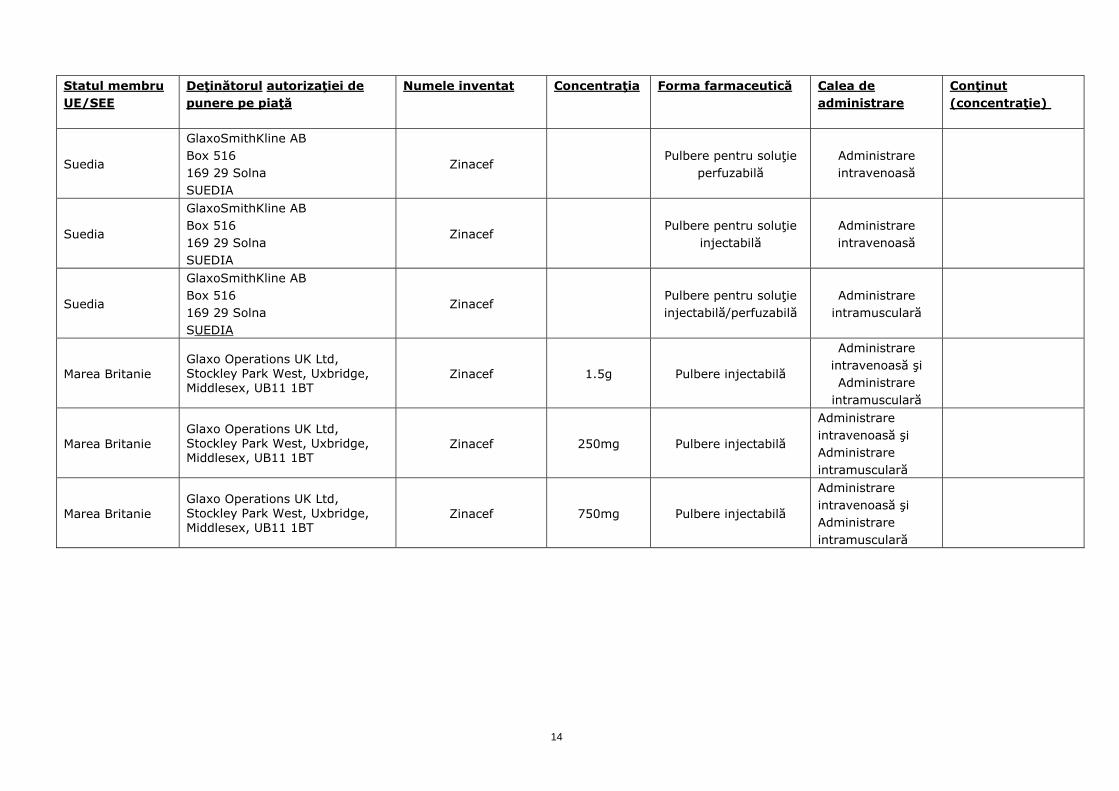
Suedia
GlaxoSmithKline AB Box 516 169 29 Solna SUEDIA
Zinacef Pulbere pentru soluţie
perfuzabilă Administrare intravenoasă
Suedia
GlaxoSmithKline AB Box 516 169 29 Solna SUEDIA
Zinacef Pulbere pentru soluţie
injectabilă Administrare intravenoasă
Suedia
GlaxoSmithKline AB Box 516 169 29 Solna SUEDIA
Zinacef Pulbere pentru soluţie injectabilă/perfuzabilă
Administrare intramusculară
Marea Britanie Glaxo Operations UK Ltd, Stockley Park West, Uxbridge, Middlesex, UB11 1BT
Zinacef 1.5g Pulbere injectabilă
Administrare intravenoasă şi Administrare
intramusculară
Marea Britanie Glaxo Operations UK Ltd, Stockley Park West, Uxbridge, Middlesex, UB11 1BT
Zinacef 250mg Pulbere injectabilă
Administrare intravenoasă şi Administrare intramusculară
Marea Britanie Glaxo Operations UK Ltd, Stockley Park West, Uxbridge, Middlesex, UB11 1BT
Zinacef 750mg Pulbere injectabilă
Administrare intravenoasă şi Administrare intramusculară

Anexa II
Concluzii ştiinţifice şi motive pentru modificarea termenilor autorizaţiilor de introducere pe piaţă
15

Concluzii ştiinţifice Rezumatul general al evaluării ştiinţifice pentru Zinacef şi denumirile asociate (vezi Anexa I) Zinacef conţine cefuroxim sodiu, un agent antibacterian de generaţia a doua din clasa cefalosporinelor. Cefuroximul are acţiune bactericidă, inhibând enzimele bacteriene necesare pentru sinteza peretelui celular (sinteza peptidoglicanului), cauzând astfel moartea celulară. Zinacef a fost aprobat pentru prima oară în Europa la începutul anilor '80 şi este disponibil ca formule parenterale. Zinacef a fost inclus în lista produselor pentru armonizarea Rezumatului Caracteristicilor Produsului (RCP), ca urmare a deciziilor divergente luate la nivel naţional de statele membre în ceea ce priveşte autorizaţia produsului menţionat mai sus. Astfel, s-a elaborat o sesizare în baza articolului 30 alineatul (2) din Directiva 2001/83/CE pentru a soluţiona aceste divergenţe şi, astfel, să se armonizeze Informaţiile despre produs (PI) pe întreg teritoriul UE. Punctul 4.1 - Indicaţii terapeutice CHMP a remarcat numărul mare de divergenţe în cadrul indicaţiilor aprobate la nivel naţional şi din acest motiv a analizat datele care sprijină fiecare indicaţie individuală şi grupele de vârstă ale pacienţilor. Pneumonie comunitară CHMP a remarcat faptul că, deşi s-a transmis doar un singur studiu dublu-orb, s-au înaintat şi alte câteva studii randomizate şi controlate cu un agent comparator, din care unele au fost realizate recent şi au demonstrat o eficacitate adecvată a cefuroximului. Astfel, CHMP a concluzionat că există suficiente date pentru a sprijini indicaţia la adulţi şi că datele eficacităţii la adulţi ar putea fi extrapolate la copii şi adolescenţi. CHMP a considerat că indicaţia este acceptabilă pentru toate populaţiile. Exacerbări acute ale bronşitei cronice CHMP a luat în considerare studiul randomizat, comparativ, dublu-orb trimis şi este de părere că acesta are o structură adecvată. Întrucât studiul a demonstrat non-inferioritatea cefuroximului, CHMP a considerat că indicaţia este acceptabilă. Infecţii ale tractului respirator superior CHMP a considerat că formularea propusă a indicaţiei este prea generală şi a remarcat faptul că majoritatea infecţiilor tractului respirator superior răspund bine la terapia orală sau se vindecă în mod spontan. CHMP a analizat studiile clinice prezentate, însă a considerat că datele sunt insuficiente. De asemenea, CHMP a observat faptul că nu există studii comparative controlate cu placebo sau dublu-oarbe în ceea ce priveşte indicaţia restrânsă pentru infecţii ORL. Astfel, CHMP a recomandat eliminarea acestei indicaţii. Infecţii ale tractului urinar CHMP a luat în considerare datele transmise, constând din unsprezece studii mici, fără comparator, şi două studii comparative, deschise. CHMP a remarcat amploarea experienţei clinice care sprijină utilizarea cefuroximului în această indicaţie. De asemenea, CHMP a remarcat faptul că există puţine opţiuni terapeutice pentru femeile gravide care au pielonefrită. În concluzie, CHMP a considerat că "infecţiile complicate de tract urinar, inclusiv pielonefrita" sunt acceptabile. Infecţii cutanate şi ale ţesutului conjunctiv moale CHMP a analizat datele transmise şi a convenit că stafilococii şi streptococii, speciile bacteriene implicate cel mai frecvent în infecţiile cutanate şi ale ţesutului conjunctiv moale, sunt sensibili la cefuroxim. Pe baza datelor furnizate, CHMP a considerat că indicaţia „infecţii ale ţesutului conjunctiv moale: celulita, erizipelul şi infecţii la nivelul plăgilor" este acceptabilă. Infecţii osoase şi articulare După analiza datelor disponibile, provenite din studii mici, fără comparator, CHMP a considerat că acestea sunt foarte limitate şi că au o metodologie îndoielnică. CHMP a considerat că datele asupra penetrării osului nu au prevalat în raport cu lipsa datelor clinice care să le sprijine. Astfel, CHMP a recomandat eliminarea acestei indicaţii. Infecţii obstetrice şi ginecologice CHMP a analizat cele două studii deschise transmise, însă a afirmat că cefuroximul nu acţionează împotriva multor specii bacteriene izolate în infecţiile obstetrice şi ginecologice, fie din cauza rezistenţei inerente, fie a rezistenţei dobândite. CHMP a considerat că această indicaţie nu este sprijinită în mod adecvat şi, astfel, a recomandat eliminarea sa.
16

Gonoree CHMP a analizat studiile transmise, dintre care majoritatea au utilizat cefuroxim în combinaţie cu probenecid, şi nu cefuroxim în monoterapie. De asemenea, CHMP a remarcat faptul că, deşi cel mai frecvent agent patogen coexistent la pacienţii cu gonoree este Chlamydia trachomatis, nu s-au transmis date asupra terapiei combinate (cefuroxim şi alt antibiotic) pentru tratamentul pacienţilor cu infecţii concomitente cu N. gonorrhoeae şi C. trachomatis sau cu N. gonorrhoeae şi bacterii anaerobe. CHMP a considerat că datele disponibile nu au sprijinit această indicaţie şi, astfel, a recomandat eliminarea sa. Septicemie şi meningită CHMP a analizat studiile asupra septicemiei, acestea fiind studii vechi, fără comparator care au cuprins un număr mic de pacienţi. Studiile au fost realizate într-o perioadă în care rezistenţa dobândită nu era o problemă critică. În ceea ce priveşte meningita, CHMP a remarcat că majoritatea studiilor au identificat H. influenzae, N. meningitidis, S. pneumonia şi S. aureus (non-MRSA) drept speciile bacteriene predominante, fapt ce nu reflectă situaţia prezentă la nivelul UE, unde bacilii gram negativi aerobi sunt agenţi cauzali din ce în ce mai importanţi. CHMP a concluzionat că datele clinice şi datele Comitetului European pentru Testarea Sensibilităţii Antimicrobiene (EUCAST) nu sprijină tratamentul meningitei. În concluzie, CHMP a considerat că datele nu sunt suficiente pentru a sprijini indicaţiile de septicemie şi meningită, şi, astfel, a recomandat eliminarea lor. Infecţii intraabdominale CHMP a analizat datele transmise şi a considerat că distribuţiile infecţiilor în cadrul celor mai mari două studii prezentate sprijină indicaţia propusă, deşi cefuroximul nu este adecvat pentru tratamentul infecţiilor cauzate de bacterii gram negative, care nu fermentează. În concluzie, CHMP a considerat că indicaţia este acceptabilă. Profilaxie După analiza tuturor datelor prezentate pentru a sprijini diversele indicaţii profilactice propuse pentru cefuroxim, CHMP a considerat că indicaţia „Profilaxia infecţiilor în cazul intervenţiilor chirurgicale gastrointestinale (inclusiv la nivelul esofagului), ortopedice, cardiovasculare şi ginecologice (inclusiv cezariană)" este acceptabilă. Indicaţia la nou-născuţi CHMP a analizat datele la nou-născuţi, inclusiv datele asupra intervalului de dozare. CHMP a definit nou-născuţii drept sugari cu vârsta mai mică de 3 săptămâni, inclusiv copiii născuţi foarte recent, şi a convenit că cefuroximul este utilizat la nou-născuţi de mulţi ani fără vreun motiv serios de îngrijorare privind siguranţa. CHMP a convenit că nou-născuţilor li se poate administra o doză zilnică totală similară cu cea administrată sugarilor (30 până la 100 mg/kg/zi), însă cu o frecvenţă zilnică redusă, egală cu 2 sau 3 doze împărţite, datorită timpului mai lung de înjumătăţire în ser. În concluzie, CHMP a adoptat următoarele indicaţii şi formulări armonizate pentru Punctul 4.1: „Zinacef este indicat pentru tratamentul infecţiilor enumerate mai jos la adulţi şi copii, inclusiv nou-născuţi (de la naştere) (vezi pct. 4.4 şi 5.1).
Pneumonie comunitară. Exacerbări acute ale bronşitei cronice. Infecţii complicate ale tractului urinar, inclusiv pielonefrită. Infecţii ale ţesutului conjunctiv moale: celulită, erizipel şi infecţii la nivelul plăgilor. Infecţii intraabdominale (vezi pct. 4.4). Profilaxia infecţiilor în cazul intervenţiilor chirurgicale gastrointestinale (inclusiv la nivelul
esofagului), ortopedice, cardiovasculare şi ginecologice (inclusiv cezariană).
În tratamentul şi prevenţia infecţiilor în care este foarte probabil să se găsească prezenţa de organisme anaerobe, cefuroximul trebuie administrat împreună cu agenţi antibacterieni adecvaţi suplimentari. Trebuie să se acorde atenţie recomandărilor oficiale privind utilizarea adecvată a agenţilor antibacterieni". Punctul 4.2 - Posologie şi mod de administrare CHMP a remarcat amploarea divergenţelor din cadrul posologiilor şi recomandărilor aprobate la nivel naţional şi, astfel, a analizat datele disponibile pentru a sprijini armonizarea Punctului 4.2. CHMP a analizat recomandările de dozaj pentru fiecare indicaţie individuală. CHMP a convenit că se prevede ca schemele de dozare pe cale intravenoasă şi intramusculară, utilizate în mod frecvent (adică, 750 mg până
17

18
la 1500 mg administrate din 8 în 8 ore), să fie eficace pentru organismele cu concentraţie inhibitorie minimă (MIC) de până la 8 μg/ml, inclusiv. CHMP a considerat că, în cazul cefuroximului administrat pe cale parenterală, bacteriile mai puţin susceptibile sunt în principal Enterobacteriaceae (adică, E. coli, P. mirabilis şi Klebsiella spp). Prin urmare, CHMP a urmat valorile critice ale EUCAST pentru Enterobacteriaceae la 8 μg/ml şi a recomandat o schemă de dozaj de 1500 mg din 8 în 8 ore pentru tratamentul infecţiilor cauzate de bacteriile de mai sus. CHMP a recomandat eliminarea opţiunii de terapie secvenţială parenterală la orală pentru toţi pacienţii, din cauza reducerii semnificative a expunerii la medicamentul activ când se trece la formula orală. Referitor la pacienţii cu insuficienţă renală, CHMP a analizat datele şi a considerat că indicaţiile de dozaj propuse sunt acceptabile. Referitor la pacienţii cu insuficienţă hepatică, CHMP a declarat că nu se aşteaptă ca disfuncţia hepatică să afecteze farmacocinetica cefuroximului. În ceea ce priveşte modul de administrare, CHMP a declarat că Zinacef trebuie administrat prin injecţie intravenoasă timp de 3 până la 5 minute, direct în venă sau prin perfuzie timp de 30 până la 60 de minute ori prin injecţie intramusculară profundă. În concluzie, CHMP a adoptat o formulare armonizată pentru Punctul 4.2. Divergenţe minore la alte puncte ale RCP-ului, etichetării şi prospectului CHMP a adoptat, de asemenea, o formulare armonizată pentru celelalte puncte ale RCP-ului pentru Zinacef şi a aliniat etichetarea şi prospectul cu RCP-ul armonizat adoptat.
Motive pentru modificarea rezumatului caracteristicilor produsului, etichetării şi prospectului
Obiectul acestei proceduri de sesizare a fost armonizarea rezumatului caracteristicilor produsului, etichetării şi prospectului. Luând în considerare datele transmise de titularul autorizaţiei de introducere pe piaţă, rapoartele de evaluare ale raportorului şi coraportorului şi discuţiile ştiinţifice din cadrul Comitetului, CHMP a fost de opinie că raportul beneficiu-risc pentru Zinacef şi denumirile asociate este favorabil.
Întrucât
Comitetul a luat în considerare sesizarea în baza articolului 30 din Directiva 2001/83/CE,
Comitetul a analizat divergenţele identificate pentru Zinacef şi denumirile asociate, referitor la
indicaţiile terapeutice şi posologia şi modul de administrare, precum şi celelalte puncte ale RCP-ului
Comitetul a analizat datele transmise de MAH, inclusiv datele provenite din studii clinice, literatura de
specialitate publicată şi documentaţia clinică, justificând Informaţiile armonizate despre produs care
au fost propuse,
Comitetul a convenit armonizarea rezumatului caracteristicilor produsului, etichetării şi prospectului,
propusă de titularii autorizaţiilor de introducere pe piaţă,
CHMP a recomandat modificarea termenilor autorizaţiilor de introducere pe piaţă, aferenţi rezumatului caracteristicilor produsului, etichetării şi prospectului din Anexa III pentru Zinacef şi denumirile asociate (vezi Anexa I).

ANEXA III
REZUMATUL CARACTERISTICILOR PRODUSULUI, ETICHETAREA ŞI PROSPECTUL
Notă: RCP-ul, etichetarea şi prospectul de faţă reprezintă versiunea valabilă în momentul adoptării deciziei Comisiei.
În urma deciziei adoptate de Comisie, autorităţile competente din statele membre, în cooperare cu statul
membru de referinţă, vor actualiza informaţiile despre produs, după caz . Prin urmare, este posibil ca RCP-ul, etichetarea şi prospectul de faţă să nu reprezinte în mod necesar textul actual.
19

REZUMATUL CARACTERISTICILOR PRODUSULUI
20

1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 500 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 1 g pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 2 g pulbere pentru soluţie perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie perfuzabilă (prezentare Monovial) Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie perfuzabilă (prezentare Monovial) [Vezi Anexa I – a se completa la nivel naţional] 2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ [A se completa la nivel naţional]
Concentraţie Zinacef Cantitatea de
sodiu per flacon
250 mg 14 mg 500 mg 28 mg 750 mg 42 mg
1 g 56 mg 1,5 g 83 mg 2 g 111 mg
3. FORMA FARMACEUTICĂ 250 mg, 750 mg, 1,5 g pulbere pentru soluţie injectabilă Pulbere pentru soluţie injectabilă [A se completa la nivel naţional] 250 mg, 500 mg, 750 mg, 1 g pulbere şi solvent pentru soluţie injectabilă Pulbere şi solvent pentru soluţie injectabilă [A se completa la nivel naţional] 250 mg, 750 mg, 1,5 g pulbere pentru soluţie injectabilă sau perfuzabilă Pulbere pentru soluţie injectabilă sau perfuzabilă [A se completa la nivel naţional] 750 mg, 1,5 g, 2 g pulbere pentru soluţie perfuzabilă Pulbere pentru soluţie perfuzabilă [A se completa la nivel naţional] 750 mg, 1,5 g pulbere pentru soluţie perfuzabilă (prezentare Monovial)
21

Pulbere pentru soluţie perfuzabilă [A se completa la nivel naţional] 4. DATE CLINICE 4.1 Indicaţii terapeutice Zinacef este indicat la adulţi şi copii, inclusiv nou-născuţi (de la naştere) pentru tratamentul infecţiilor enumerate mai jos (vezi punctele 4.4 şi 5.1). Pneumonie comunitară dobândită. Exacerbări acute ale bronşitei cronice. Infecţii ale tractului urinar complicate, incluzând pielonefrite. Infecţii ale ţesuturilor moi: celulită, erizipel şi plăgi infectate Infecţii intra-abdominale (vezi punctul 4.4) Profilaxia infecţiilor în chirurgia gastro-intestinală (inclusiv esofagiană), ortopedică, cardiovasculară,
ginecologică (inclusiv operaţii cezariene).
În tratamentul şi prevenirea infecţiilor în care este foarte probabil fie implicate microorganisme anaerobe, cefuroxima trebuie administrată în asociere cu substanţe antibacteriene adecvate. Trebuie avute în vedere ghidurile terapeutice în vigoare cu privire la utilizarea adecvată a antibioticelor. 4.2 Doze şi mod de administrare Doze Tabelul 1. Adulţi, adolescenţi şi copii cu greutatea 40 kg
Indicaţie Doză Pneumonie comunitară dobândită şi exacerbări acute ale bronşitei cronice. Infecţii ale ţesuturilor moi: celulită, erizipel şi plăgi infectate. Infecţii intra-abdominale
750 mg la interval de 8 ore (intravenos sau intramuscular)
Infecţii ale tractului urinar complicate, incluzând pielonefrite
1,5 g la interval de 8 ore (intravenos sau intramuscular)
Infecţii severe 750 mg la interval de 6 ore (intravenos) 1,5 g la interval de 8 ore (intravenos)
Profilaxia infecţiilor în chirurgia gastro-intestinală, ginecologică (inclusiv operaţii cezariene) şi ortopedică.
1,5 g la inducerea anesteziei. Această doză se poate suplimenta cu două doze a câte 750 mg (intramuscular) după 8 ore şi ulterior după 16 ore.
22
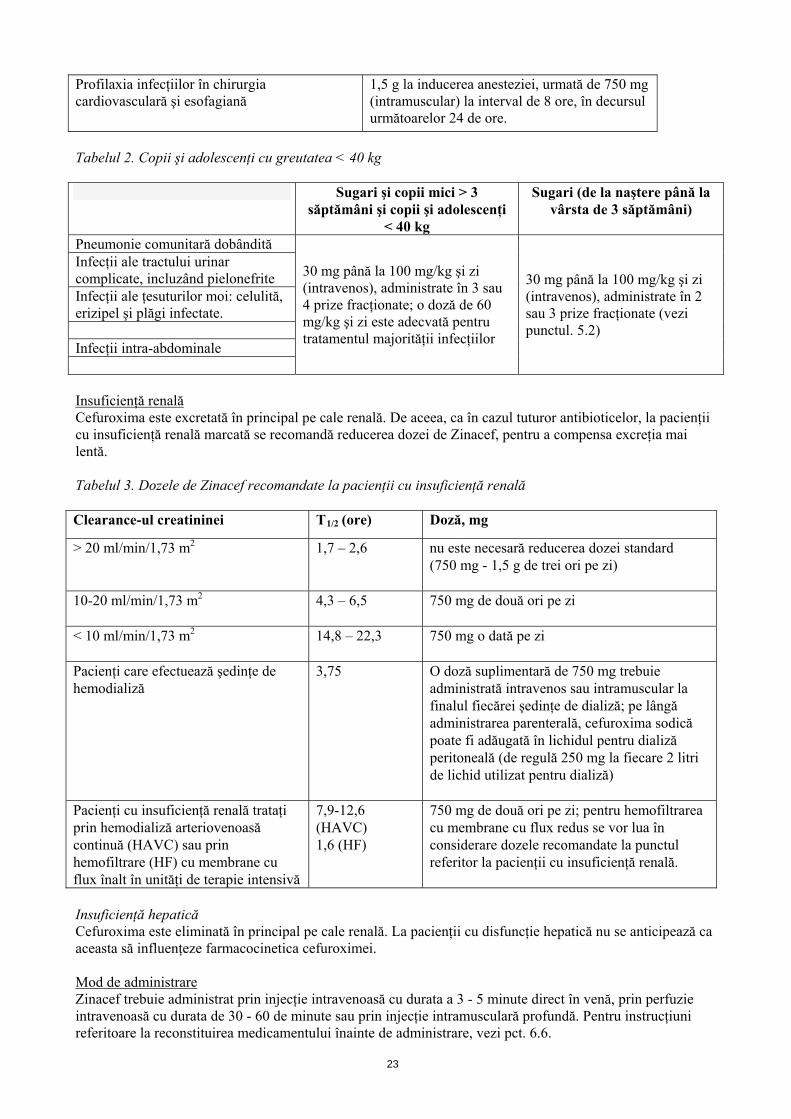
Profilaxia infecţiilor în chirurgia cardiovasculară şi esofagiană
1,5 g la inducerea anesteziei, urmată de 750 mg (intramuscular) la interval de 8 ore, în decursul următoarelor 24 de ore.
Tabelul 2. Copii şi adolescenţi cu greutatea < 40 kg Sugari şi copii mici > 3
săptămâni şi copii şi adolescenţi < 40 kg
Sugari (de la naştere până la vârsta de 3 săptămâni)
Pneumonie comunitară dobândită Infecţii ale tractului urinar complicate, incluzând pielonefrite Infecţii ale ţesuturilor moi: celulită, erizipel şi plăgi infectate. Infecţii intra-abdominale
30 mg până la 100 mg/kg şi zi (intravenos), administrate în 3 sau 4 prize fracţionate; o doză de 60 mg/kg şi zi este adecvată pentru tratamentul majorităţii infecţiilor
30 mg până la 100 mg/kg şi zi (intravenos), administrate în 2 sau 3 prize fracţionate (vezi punctul. 5.2)
Insuficienţă renală Cefuroxima este excretată în principal pe cale renală. De aceea, ca în cazul tuturor antibioticelor, la pacienţii cu insuficienţă renală marcată se recomandă reducerea dozei de Zinacef, pentru a compensa excreţia mai lentă. Tabelul 3. Dozele de Zinacef recomandate la pacienţii cu insuficienţă renală Clearance-ul creatininei T1/2 (ore) Doză, mg
> 20 ml/min/1,73 m2 1,7 – 2,6 nu este necesară reducerea dozei standard (750 mg - 1,5 g de trei ori pe zi)
10-20 ml/min/1,73 m2 4,3 – 6,5 750 mg de două ori pe zi
< 10 ml/min/1,73 m2 14,8 – 22,3 750 mg o dată pe zi
Pacienţi care efectuează şedinţe de hemodializă
3,75 O doză suplimentară de 750 mg trebuie administrată intravenos sau intramuscular la finalul fiecărei şedinţe de dializă; pe lângă administrarea parenterală, cefuroxima sodică poate fi adăugată în lichidul pentru dializă peritoneală (de regulă 250 mg la fiecare 2 litri de lichid utilizat pentru dializă)
Pacienţi cu insuficienţă renală trataţi prin hemodializă arteriovenoasă continuă (HAVC) sau prin hemofiltrare (HF) cu membrane cu flux înalt în unităţi de terapie intensivă
7,9-12,6 (HAVC) 1,6 (HF)
750 mg de două ori pe zi; pentru hemofiltrarea cu membrane cu flux redus se vor lua în considerare dozele recomandate la punctul referitor la pacienţii cu insuficienţă renală.
Insuficienţă hepatică Cefuroxima este eliminată în principal pe cale renală. La pacienţii cu disfuncţie hepatică nu se anticipează ca aceasta să influenţeze farmacocinetica cefuroximei. Mod de administrare Zinacef trebuie administrat prin injecţie intravenoasă cu durata a 3 - 5 minute direct în venă, prin perfuzie intravenoasă cu durata de 30 - 60 de minute sau prin injecţie intramusculară profundă. Pentru instrucţiuni referitoare la reconstituirea medicamentului înainte de administrare, vezi pct. 6.6.
23

750 mg, 1,5 g pulbere pentru soluţie perfuzabilă (prezentare Monovial). Pentru instrucţiuni referitoare la prepararea medicamentului înainte de administrare, vezi pct. 6.6. 4.3 Contraindicaţii Hipersensibilitate la cefuroximă sau la oricare dintre excipienţii listaţi la punctul 6.1. Pacienţi cu hipersensibilitate cunoscută la alte antibiotice din clasa cefalosporinelor. Antecedente de hipersensibilitate severă (de exemplu, reacţii anafilactice) la orice alt tip de medicament antibacterian betalactamic (peniciline, monobactami şi carbapeneme). 4.4 Atenţionări şi precauţii speciale pentru utilizare Reacţii de hipersensibilitate Ca şi în cazul tuturor antibioticelor beta-lactamice, au fost raportate reacţii de hipersensibilitate grave şi ocazional letale. În cazul apariţiei reacţiilor de hipersensibilitate severe, tratamentul cu cefuroximă trebuie întrerupt imediat şi trebuie iniţiate măsuri de urgenţă adecvate. Înainte de începerea tratamentului, trebuie să se stabilească dacă pacientul are antecedente de reacţii de hipersensibilitate severe la cefuroximă, la alte cefalosporine sau la oricare alt tip de antibiotic beta-lactamic. Cefuroxima trebuie administrată cu precauţie la pacienţii cu antecedente de reacţii de hipersensibilitate non-severe la alte antibiotice betalactamice. Administrarea concomitentă cu diuretice potente sau cu aminoglicozide La pacienţii trataţi concomitent cu diuretice potente, cum este furosemidul sau cu aminoglicozide, administrarea antibioticelor din clasa cefalosporinelor în doze mari trebuie efectuată cu precauţie. Insuficienţa renală a fost raportată în timpul utilizării concomitente a acestor substanţe. Funcţia renală trebuie monitorizată la vârstnici şi la pacienţii cu insuficienţă renală pre-existentă diagnosticată (vezi punctul 4.2). Dezvoltarea microorganismelor non-susceptibile la tratament Terapia cu cefuroximă poate determina dezvoltarea excesivă a microorganismelor de tip Candida. Utilizarea prelungită poate determina şi dezvoltarea excesivă a altor microorganisme non-susceptibile la tratament (de exemplu, enterococi şi Clostridium difficile), fapt care poate impune întreruperea tratamentului (vezi punctul 4.8). Cazuri de colită pseudomembranoasă asociată antibioterapiei au fost raportate în cazul tratamentului cu cefuroximă şi aceasta poate avea grade diferite de severitate, de la uşoară până la forme care pot pune viaţa în pericol. Acest diagnostic trebuie avut în vedere la pacienţii cu diaree apărută în timpul sau după administrarea de cefuroximă (vezi punctul 4.8). Trebuie luată în considerare întreruperea terapiei cu cefuroximă şi administrarea tratamentului specific pentru Clostridium difficile. Nu trebuie administrate medicamente care inhibă peristaltismul intestinal. Infecţii intra-abdominale Având în vedere spectrul de activitate al cefuroximei, aceasta nu este adecvată pentru tratamentul infecţiilor cauzate de bacterii Gram-negativ, care nu fermentează (vezi punctul 5.1) Interferenţă cu investigaţiile diagnostice Pozitivarea rezultatelor testului Coombs asociată utilizării cefuroximei poate interfera cu testele de compatibilitate sanguină (vezi punctul 4.8). Se pot observa interferenţe uşoare cu metodele de reducere a cuprului (Benedict, Fehling, Clinitest). În orice caz, acest lucru nu duce la rezultate fals-pozitive, cum se întâmplă în cazul altor cefalosporine.
24

Deoarece în cazul testului cu fericianură pot apărea rezultate fals negative, se recomandă să se utilizeze fie metoda glucozo-oxidazei fie cea cu hexochinază pentru determinarea valorilor glucozei din sânge/plasmă la pacienţii trataţi cu cefuroximă sodică. Informaţii importante despre excipienţi Zinacef pulbere pentru soluţie injectabilă şi perfuzabilă conţine sodiu. Acest aspect trebuie avut în vedere la pacienţii care urmează o dietă cu restricţie de sodiu. 4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune Cefuroxima poate avea efecte asupra microflorei intestinale, determinând o reabsorbţie scăzută a estrogenului şi reducerea eficacităţii contraceptivelor orale combinate. Cefuroxima se excretă prin filtrare glomerulară şi secreţie tubulară. Administrarea concomitentă cu probenecid nu este recomandată. Administrarea concomitentă cu probenecid determină prelungirea perioadei de excreţie a antibioticului şi creşterea concentraţiilor plasmatice ale acestuia. Medicamente cu potenţial nefrotoxic şi diuretice de ansă La pacienţii care utilizează diuretice cu acţiune puternică (cum este furosemid) sau medicamente cu potenţial nefrotoxic (cum sunt antibioticele aminoglicozide), administrarea concomitentă de doze mari de cefalosporine trebuie efectuată cu precauţie, deoarece nu poate fi exclusă insuficienţa funcţiei renale în cazul utilizării concomitente a unor astfel de substanţe. Alte interacţiuni Determinarea concentraţiei de glucoză din sânge/plasmă: vă rugăm să citiţi punctul 4.4. Utilizarea concomitentă cu anticoagulantele orale poate determina creşterea valorilor INR. 4.6 Fertilitatea, sarcina şi alăptarea Sarcina Datele provenite din utilizarea cefuroximei la femeile gravide sunt limitate. Studiile la animale nu au evidenţiat toxicitate asupra funcţiei de reproducere (vezi punctul 5.3). Zinacef trebuie prescris la femeile gravide doar dacă beneficiile depăşesc eventualele riscuri. S-a demonstrat că cefuroxima traversează placenta şi, după administrarea intramusculară sau intravenoasă a dozei la mamă, atinge concentraţii terapeutice în lichidul amniotic şi la nivelul cordonului ombilical. Alăptarea Cefuroxima se excretă în cantităţi mici în laptele uman. Nu se anticipează aparaţia reacţiilor adverse în cazul administrării dozelor terapeutice, deşi nu se poate exclude riscul de apariţie a diareei şi a infecţiilor fungice la nivelul mucoaselor. Este posibil să fie necesară luarea unei decizii cu privire la întreruperea alăptării sau la întreruperea tratamentului cu cefuroximă, luând în considerare beneficiul alăptării pentru copil şi beneficiul tratamentului pentru mamă. Fertilitatea Nu sunt disponibile date referitoare la efectele cefuroximei sodice asupra fertilităţii la om. Studiile privind toxicitatea asupra funcţiei de reproducere efectuate la animale nu au evidenţiat efecte asupra fertilităţii. 4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje Nu s-au efectuat studii referitoare la efectele asupra capacităţii de a conduce vehicule şi de a folosi utilaje. Cu toate acestea, pe baza reacţiilor adverse cunoscute, este puţin probabil că cefuroxima să afecteze capacitatea de a conduce vehicule şi de a folosi utilaje.
25
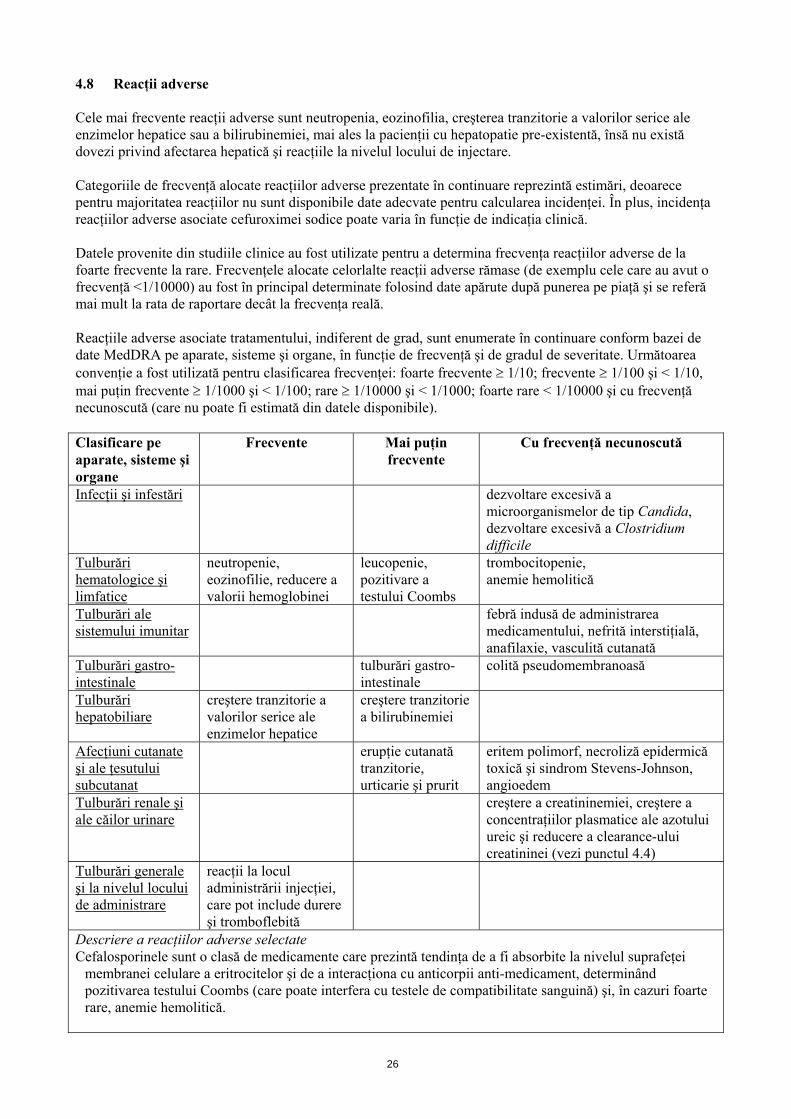
4.8 Reacţii adverse Cele mai frecvente reacţii adverse sunt neutropenia, eozinofilia, creşterea tranzitorie a valorilor serice ale enzimelor hepatice sau a bilirubinemiei, mai ales la pacienţii cu hepatopatie pre-existentă, însă nu există dovezi privind afectarea hepatică şi reacţiile la nivelul locului de injectare. Categoriile de frecvenţă alocate reacţiilor adverse prezentate în continuare reprezintă estimări, deoarece pentru majoritatea reacţiilor nu sunt disponibile date adecvate pentru calcularea incidenţei. În plus, incidenţa reacţiilor adverse asociate cefuroximei sodice poate varia în funcţie de indicaţia clinică. Datele provenite din studiile clinice au fost utilizate pentru a determina frecvenţa reacţiilor adverse de la foarte frecvente la rare. Frecvenţele alocate celorlalte reacţii adverse rămase (de exemplu cele care au avut o frecvenţă <1/10000) au fost în principal determinate folosind date apărute după punerea pe piaţă şi se referă mai mult la rata de raportare decât la frecvenţa reală. Reacţiile adverse asociate tratamentului, indiferent de grad, sunt enumerate în continuare conform bazei de date MedDRA pe aparate, sisteme şi organe, în funcţie de frecvenţă şi de gradul de severitate. Următoarea convenţie a fost utilizată pentru clasificarea frecvenţei: foarte frecvente 1/10; frecvente 1/100 şi < 1/10, mai puţin frecvente 1/1000 şi < 1/100; rare 1/10000 şi < 1/1000; foarte rare < 1/10000 şi cu frecvenţă necunoscută (care nu poate fi estimată din datele disponibile). Clasificare pe aparate, sisteme şi organe
Frecvente Mai puţin frecvente
Cu frecvenţă necunoscută
Infecţii şi infestări dezvoltare excesivă a microorganismelor de tip Candida, dezvoltare excesivă a Clostridium difficile
Tulburări hematologice şi limfatice
neutropenie, eozinofilie, reducere a valorii hemoglobinei
leucopenie, pozitivare a testului Coombs
trombocitopenie, anemie hemolitică
Tulburări ale sistemului imunitar
febră indusă de administrarea medicamentului, nefrită interstiţială, anafilaxie, vasculită cutanată
Tulburări gastro-intestinale
tulburări gastro-intestinale
colită pseudomembranoasă
Tulburări hepatobiliare
creştere tranzitorie a valorilor serice ale enzimelor hepatice
creştere tranzitorie a bilirubinemiei
Afecţiuni cutanate şi ale ţesutului subcutanat
erupţie cutanată tranzitorie, urticarie şi prurit
eritem polimorf, necroliză epidermică toxică şi sindrom Stevens-Johnson, angioedem
Tulburări renale şi ale căilor urinare
creştere a creatininemiei, creştere a concentraţiilor plasmatice ale azotului ureic şi reducere a clearance-ului creatininei (vezi punctul 4.4)
Tulburări generale şi la nivelul locului de administrare
reacţii la locul administrării injecţiei, care pot include durere şi tromboflebită
Descriere a reacţiilor adverse selectate Cefalosporinele sunt o clasă de medicamente care prezintă tendinţa de a fi absorbite la nivelul suprafeţei
membranei celulare a eritrocitelor şi de a interacţiona cu anticorpii anti-medicament, determinând pozitivarea testului Coombs (care poate interfera cu testele de compatibilitate sanguină) şi, în cazuri foarte rare, anemie hemolitică.
26

Au fost observate creşteri tranzitorii ale valorilor serice ale enzimelor hepatice sau ale bilirubinemiei, de regulă reversibile.
Durerea la locul injectării intramusculare este mai probabil să apară în cazul administrării unor doze mai
mari. Cu toate acestea, este puţin probabil ca acest lucru să determine întreruperea tratamentului. Copii şi adolescenţi Profilul de siguranţă al cefuroximei sodice la copii şi adolescenţi este concordant cu profilul de siguranţă observat la adulţi. 4.9 Supradozaj Supradozajul poate determina sechele neurologice, incluzând encefalopatie, convulsii şi comă. Simptomele supradozajului pot apărea în cazul în care doza nu se reduce adecvat la pacienţii cu insuficienţă renală (vezi punctele 4.2 şi 4.4). Concentraţiile plasmatice ale cefuroximei pot fi reduse prin hemodializă şi dializă peritoneală. 5. PROPRIETĂŢI FARMACOLOGICE 5.1 Proprietăţi farmacodinamice Grupa farmacoterapeutică: antibiotice pentru administrare sistemică, cefalosporine de generaţia a doua, codul ATC: J01DC02 Mecanism de acţiune Cefuroxima inhibă sinteza peretelui bacterian, după legarea de proteinele de legare a penicilinei (PLP). Astfel se întrerupe biosinteza peretelui celular (peptidoglican), fapt care determină liza şi apoptoza celulelor bacteriene. Mecanism de rezistenţă Rezistenţa bacteriană la cefuroximă poate fi determinată de unul sau mai multe dintre următoarele mecanisme: hidroliza de către beta-lactamaze incluzând (dar nu linitându-se la) beta-lactamazele cu spectru extins
(BLSE) şi enzimele Amp-C, a căror expresie ar putea fi indusă sau inhibată stabil la anumite specii bacteriene aerobe Gram-negativ;
afinitatea redusă a proteinelor de legare a penicilinei pentru cefuroximă; lipsa de permeabilitate a membranei externe, care restricţionează accesul cefuroximei la nivelul
proteinelor de legare a penicilinei în cazul bacteriilor Gram-negativ; pompele bacteriene de eflux.
Este de aşteptat ca microorganismele care au dobândit rezistenţă la alte cefalosporine administrate injectabil să fie rezistente la cefuroximă. În funcţie de mecanismul de rezistenţă, microorganismele cu rezistenţă dobândită la penicilină pot prezenta sensibilitate scăzută sau rezistenţă la cefuroximă.
Valori critice pentru cefuroximă sodică Valorile critice ale CMI (concentraţia minimă inhibitorie) stabilite de EUCAST (European Committee on Antimicrobial Susceptibility Testing) sunt prezentate în continuare:
Microorganism Valori critice (mg/l)
27
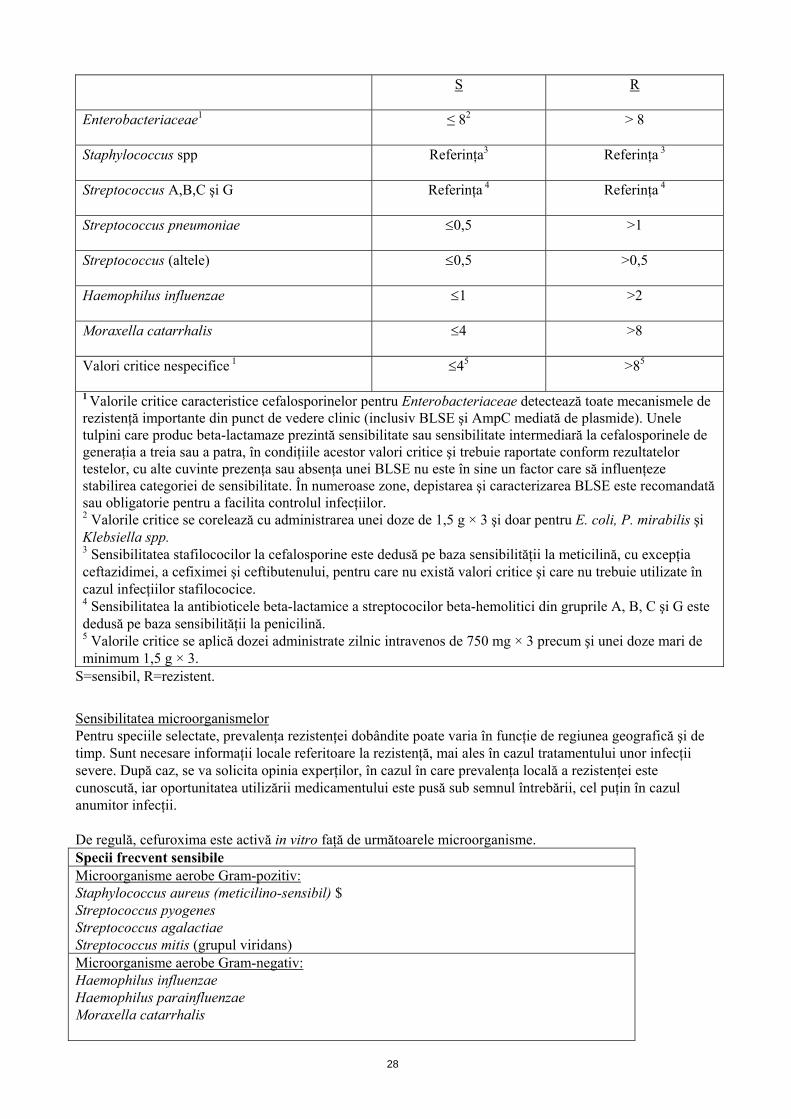
S R
Enterobacteriaceae1 ≤ 82 > 8
Staphylococcus spp Referinţa3 Referinţa 3
Streptococcus A,B,C şi G Referinţa 4 Referinţa 4
Streptococcus pneumoniae 0,5 >1
Streptococcus (altele) 0,5 >0,5
Haemophilus influenzae 1 >2
Moraxella catarrhalis 4 >8
Valori critice nespecifice 1 45 >85
1 Valorile critice caracteristice cefalosporinelor pentru Enterobacteriaceae detectează toate mecanismele de rezistenţă importante din punct de vedere clinic (inclusiv BLSE şi AmpC mediată de plasmide). Unele tulpini care produc beta-lactamaze prezintă sensibilitate sau sensibilitate intermediară la cefalosporinele de generaţia a treia sau a patra, în condiţiile acestor valori critice şi trebuie raportate conform rezultatelor testelor, cu alte cuvinte prezenţa sau absenţa unei BLSE nu este în sine un factor care să influenţeze stabilirea categoriei de sensibilitate. În numeroase zone, depistarea şi caracterizarea BLSE este recomandată sau obligatorie pentru a facilita controlul infecţiilor. 2 Valorile critice se corelează cu administrarea unei doze de 1,5 g × 3 şi doar pentru E. coli, P. mirabilis şi Klebsiella spp. 3 Sensibilitatea stafilococilor la cefalosporine este dedusă pe baza sensibilităţii la meticilină, cu excepţia ceftazidimei, a cefiximei şi ceftibutenului, pentru care nu există valori critice şi care nu trebuie utilizate în cazul infecţiilor stafilococice. 4 Sensibilitatea la antibioticele beta-lactamice a streptococilor beta-hemolitici din gruprile A, B, C şi G este dedusă pe baza sensibilităţii la penicilină. 5 Valorile critice se aplică dozei administrate zilnic intravenos de 750 mg × 3 precum şi unei doze mari de minimum 1,5 g × 3.
S=sensibil, R=rezistent.
Sensibilitatea microorganismelor Pentru speciile selectate, prevalenţa rezistenţei dobândite poate varia în funcţie de regiunea geografică şi de timp. Sunt necesare informaţii locale referitoare la rezistenţă, mai ales în cazul tratamentului unor infecţii severe. După caz, se va solicita opinia experţilor, în cazul în care prevalenţa locală a rezistenţei este cunoscută, iar oportunitatea utilizării medicamentului este pusă sub semnul întrebării, cel puţin în cazul anumitor infecţii. De regulă, cefuroxima este activă in vitro faţă de următoarele microorganisme. Specii frecvent sensibile Microorganisme aerobe Gram-pozitiv: Staphylococcus aureus (meticilino-sensibil) $ Streptococcus pyogenes Streptococcus agalactiae Streptococcus mitis (grupul viridans) Microorganisme aerobe Gram-negativ: Haemophilus influenzae Haemophilus parainfluenzae Moraxella catarrhalis
28

Microorganisme în cazul cărora rezistenţa ar putea reprezenta o problemă Microorganisme aerobe Gram-pozitiv: Streptococcus pneumoniae Microorganisme aerobe Gram-negativ: Citrobacter freundii Enterobacter cloacae Enterobacter aerogenes Escherichia coli Klebsiella pneumoniae Proteus mirabilis Proteus spp. (altele decât P. vulgaris) Providencia spp. Salmonella spp. Microorganisme anaerobe Gram-pozitiv: Peptostreptococcus spp. Propionibacterium spp. Microorganisme anaerobe Gram-negativ: Fusobacterium spp. Bacteroides spp. Microorganisme cu rezistenţă inerentă Microorganisme aerobe Gram-pozitiv: Enterococcus faecalis Enterococcus faecium Microorganisme aerobe Gram-negativ: Acinetobacter spp Morganella morganii Proteus vulgaris Pseudomonas aeruginosa Serratia marcescens Microorganisme anaerobe Gram-pozitiv: Clostridium difficile Microorganisme anaerobe Gram-negativ: Bacteroides fragilis Altele: Chlamydia spp Mycoplasma spp Legionella spp $ Toate speciile de S. aureus meticilino-rezistente sunt rezistente la cefuroximă.
S-a demonstrat că activitatea in vitro a cefuroximei sodice şi a antibioticelor aminoglicozidice administrate în asociere este cel puţin aditivă, evidenţiindu-se ocazional activitate sinergică.
5.2 Proprietăţi farmacocinetice Absorbţie După injectarea intramusculară (IM) a cefuroximei la voluntari sănătoşi, valoarea medie a concentraţiilor plasmatice maxime a variat între 27 şi 35 µg/ml pentru o doză de 750 mg şi între 33 şi 40 µg/ml pentru o doză de 1000 mg, valorile fiind obţinute în decurs de 30 - 60 de minute după administrare. După 15 minute de la administrarea intravenoasă (i.v.) a dozelor de 750 şi 1500 mg, concentraţiile plasmatice au fost de aproximativ 50 µg/ml şi, respectiv de 100 µg/ml. După administrarea i.m. şi i.v., se pare că ASC şi Cmax cresc liniar o dată cu creşterea dozei, în intervalul de doze unice terapeutice cuprinse între 250 şi 1000 mg. Nu s-a evidenţiat acumularea cefuroximei în probele
29

de ser prelevate de la voluntari sănătoşi după administrarea intravenoasă repetată a unor doze de 1500 mg la interval de 8 ore. Distribuţie S-a constatat că legarea de proteinele plasmatice se face în proporţie de 33 - 50%, în funcţie de metoda folosită. Volumul mediu de distribuţie variază între 9,3 – 15,8 l/1,73 m2 după administrarea i.m.sau i.v., în intervalul de doze terapeutice cuprinse între 250 şi 1000 mg. Concentraţii ale cefuroximei mai mari decât valorile inhibitorii minime pentru microorganismele patogene frecvent întânite pot fi obţinute în ţesuturile de la nivelul amigdalelor, sinusurilor, în mucoasa bronşică, în os, lichid pleural, lichid articular, lichid sinovial, lichid interstiţial, bilă, spută şi umoarea apoasă. Cefuroxima traversează bariera hematoencefalică în cazul inflamaţiei meningelui. Biotransformare Cefuroxima nu este metabolizată. Eliminare Cefuroxima este excretată prin filtrare glomerulară şi secreţie tubulară. Timpul de înjumătăţire plasmatică după administrarea injectabilă intramusculară sau intravenoasă este de aproximativ 70 de minute. Cefuroxima se recuperează aproape complet (85 - 90%) sub formă de cefuroximă nemodificată în urină, în interval de 24 ore după administrare. Cea mai mare parte din doza de cefuroximă administrată se excretă în primele 6 ore. Valorile medii ale clearance-ului renal variază între 114 şi 170 ml/min/1,73 m2 după administrarea i.m. sau i.v., în intervalul de doze terapeutice cuprinse între 250 - 1000 mg. Grupe speciale de pacienţi Sex Nu s-au observat diferenţe în ceea ce priveşte farmacocinetica cefuroximei între bărbaţi şi femei, după administrarea unică în bolus i.v. a 1000 mg cefuroximă sub formă de sare sodică. Vârstnici După administrarea i.m. sau i.v., absorbţia, distribuţia şi excreţia cefuroximei la pacienţii vârstnici sunt similare cu cele observate la pacienţii mai tineri cu funcţie renală echivalentă. Deoarece pacienţii vârstnici au o probabilitate mai mare de a prezenta reducere a funcţiei renale, se recomandă precauţie atunci când se stabileşte doza de cefuroximă, iar monitorizarea funcţiei renale poate fi utilă (vezi punctul 4.2). Copii şi adolescenţi S-a demonstrat că timpul de înjumătăţire plasmatică a cefuroximei este semnificativ prelungit la nou-născuţi, în funcţie de vârsta gestaţională. Cu toate acestea, la sugarii mai mari (cu vârsta >3 săptămâni), la copii şi adolescenţi, timpul de înjumătăţire plasmatică cuprins între 60 şi 90 de minute este similar celui observat la adulţi. Insuficienţă renală Cefuroxima se excretă în principal pe cale renală. Ca în cazul celorlalte antibiotice similare, la pacienţii cu insuficienţă renală marcată (C1cr < 20 ml/minut) se recomandă reducerea dozei de cefuroximă, pentru a compensa excreţia mai lentă a acesteia (vezi punctul 4.2). Cefuroxima este eliminată eficient prin hemodializă şi dializă peritoneală. Insuficienţă hepatică
30

Deoarece cefuroxima este eliminată în principal pe cale renală, nu se aşteaptă ca disfuncţia hepatică să influenţeze farmacocinetica cefuroximei. Relaţia farmacocinetică/farmacodinamie Pentru cefalosporine, s-a demonstrat că cel mai important parametru de farmacocinetică-farmacodinamie corelat cu eficacitatea in vivo este proporţia din intervalul de administrare (%T) în care concentraţia nelegată se menţine peste concentraţia minimă inhibitorie (CMI) a cefuroximei pentru speciile ţintă selectate (%T>CMI). 5.3 Date preclinice de siguranţă Datele non-clinice nu au evidenţiat niciun risc special pentru om pe baza studiilor farmacologice privind evaluarea siguranţei, toxicitatea după doze repetate, genotoxicitatea şi toxicitatea asupra funcţiei de reproducere şi dezvoltării. Nu s-au efectuat studii privind carcinogenitatea; cu toate acestea, dovezile existente nu indică potenţial carcinogen. Activitatea gama glutamil transpeptidazei în urina de şobolan este inhibată de o serie de cefalosporine, însă nivelul de inhibare este mai mic în cazul cefuroximei. Acest aspect ar putea fi relevant pentru interferenţa cu testele paraclinice la om. 6. PROPRIETĂŢI FARMACEUTICE 6.1 Lista excipienţilor [A se completa la nivel naţional] 6.2 Incompatibilităţi [A se completa la nivel naţional] 6.3 Perioada de valabilitate [A se completa la nivel naţional] 6.4 Precauţii speciale pentru păstrare [A se completa la nivel naţional] 6.5 Natura şi conţinutul ambalajului [A se completa la nivel naţional] 6.6 Precauţii speciale pentru eliminarea reziduurilor şi alte instrucţiuni de manipulare Instrucţiuni de reconstituire Tabelul 4. Volumul care trebuie adăugat şi concentraţiile soluţiei obţinute, de utilizat în cazul în care este necesară administrarea de doze fracţionate.
Volumul care trebuie adăugat şi concentraţiile soluţiei obţinute, de utilizat în cazul în care este necesară administrarea de doze fracţionate
Volumul flaconului Cantitatea de apă care trebuie adăugată (ml)
Concentraţia aproximativă a
cefuroximei (mg/ml)**
31
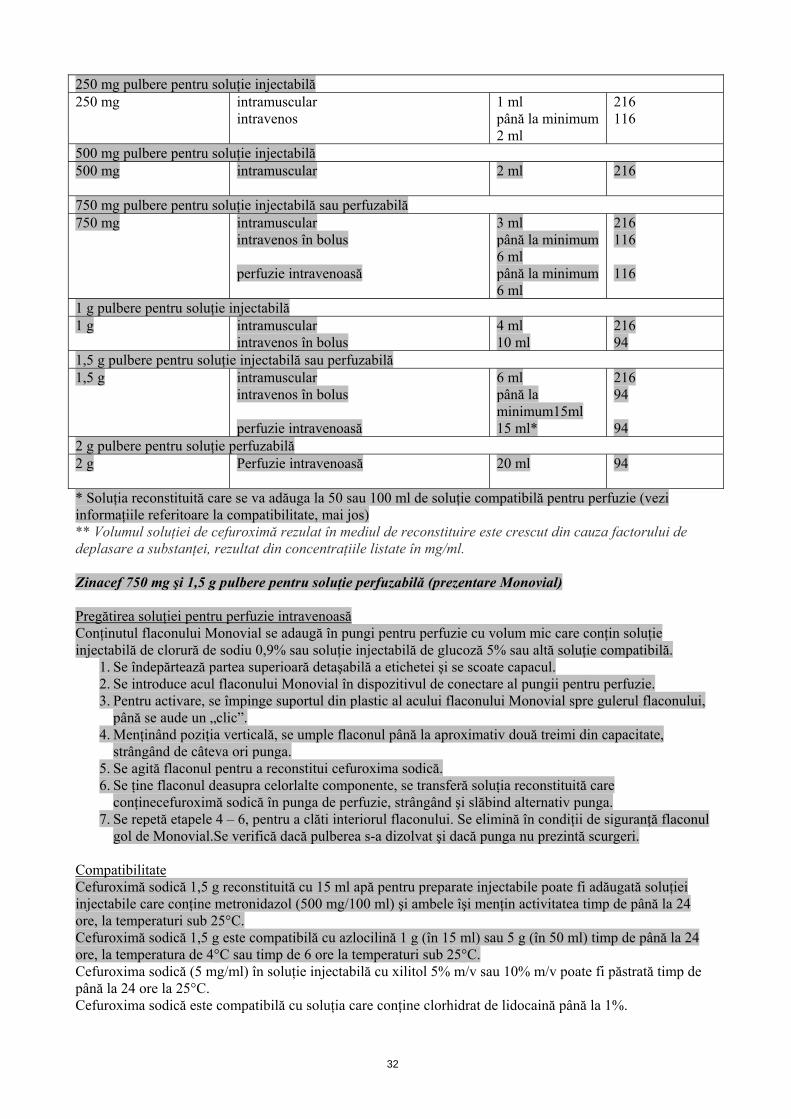
250 mg pulbere pentru soluţie injectabilă 250 mg intramuscular
intravenos 1 ml până la minimum 2 ml
216 116
500 mg pulbere pentru soluţie injectabilă 500 mg intramuscular
2 ml 216
750 mg pulbere pentru soluţie injectabilă sau perfuzabilă 750 mg intramuscular
intravenos în bolus perfuzie intravenoasă
3 ml până la minimum 6 ml până la minimum 6 ml
216 116 116
1 g pulbere pentru soluţie injectabilă 1 g
intramuscular intravenos în bolus
4 ml 10 ml
216 94
1,5 g pulbere pentru soluţie injectabilă sau perfuzabilă 1,5 g
intramuscular intravenos în bolus perfuzie intravenoasă
6 ml până la minimum15ml 15 ml*
216 94 94
2 g pulbere pentru soluţie perfuzabilă 2 g
Perfuzie intravenoasă 20 ml 94
* Soluţia reconstituită care se va adăuga la 50 sau 100 ml de soluţie compatibilă pentru perfuzie (vezi informaţiile referitoare la compatibilitate, mai jos) ** Volumul soluţiei de cefuroximă rezulat în mediul de reconstituire este crescut din cauza factorului de deplasare a substanţei, rezultat din concentraţiile listate în mg/ml. Zinacef 750 mg şi 1,5 g pulbere pentru soluţie perfuzabilă (prezentare Monovial) Pregătirea soluţiei pentru perfuzie intravenoasă Conţinutul flaconului Monovial se adaugă în pungi pentru perfuzie cu volum mic care conţin soluţie injectabilă de clorură de sodiu 0,9% sau soluţie injectabilă de glucoză 5% sau altă soluţie compatibilă.
1. Se îndepărtează partea superioară detaşabilă a etichetei şi se scoate capacul. 2. Se introduce acul flaconului Monovial în dispozitivul de conectare al pungii pentru perfuzie. 3. Pentru activare, se împinge suportul din plastic al acului flaconului Monovial spre gulerul flaconului,
până se aude un „clic”. 4. Menţinând poziţia verticală, se umple flaconul până la aproximativ două treimi din capacitate,
strângând de câteva ori punga. 5. Se agită flaconul pentru a reconstitui cefuroxima sodică. 6. Se ţine flaconul deasupra celorlalte componente, se transferă soluţia reconstituită care
conţinecefuroximă sodică în punga de perfuzie, strângând şi slăbind alternativ punga. 7. Se repetă etapele 4 – 6, pentru a clăti interiorul flaconului. Se elimină în condiţii de siguranţă flaconul
gol de Monovial.Se verifică dacă pulberea s-a dizolvat şi dacă punga nu prezintă scurgeri. Compatibilitate Cefuroximă sodică 1,5 g reconstituită cu 15 ml apă pentru preparate injectabile poate fi adăugată soluţiei injectabile care conţine metronidazol (500 mg/100 ml) şi ambele îşi menţin activitatea timp de până la 24 ore, la temperaturi sub 25°C. Cefuroximă sodică 1,5 g este compatibilă cu azlocilină 1 g (în 15 ml) sau 5 g (în 50 ml) timp de până la 24 ore, la temperatura de 4°C sau timp de 6 ore la temperaturi sub 25°C. Cefuroxima sodică (5 mg/ml) în soluţie injectabilă cu xilitol 5% m/v sau 10% m/v poate fi păstrată timp de până la 24 ore la 25°C. Cefuroxima sodică este compatibilă cu soluţia care conţine clorhidrat de lidocaină până la 1%.
32

Cefuroxima sodică este compatibilă cu următoarele soluţii perfuzabile. Îşi va menţine potenţa timp de până la 24 ore, la temperatura camerei în:
Clorură de sodiu 0,9% m/v soluţie pentru preparate injectabile BP Glucoză 5% soluţie pentru preparate injectabile BP Clorură de sodiu 0,18% m/v plus glucoză 4% soluţie pentru preparate injectabile BP Glucoză 5% şi clorură de sodiu 0,9% soluţie pentru preparate injectabile Glucoză 5% şi clorură de sodiu 0,45% soluţie pentru preparate injectabile Glucoză 5% şi clorură de sodiu 0,225% soluţie pentru preparate injectabile Glucoză 10% soluţie pentru preparate injectabile Zahăr invertit 10% în apă pentru preparate injectabile Soluţie injectabilă Ringer USP Soluţie injectabilă Ringer lactat USP Soluţie injectabilă de lactat de sodiu M/6 Soluţie injectabilă de lactat de sodiu compus BP (soluţie Hartmann).
Stabilitatea cefuroximei sodice în soluţie pentru preparate injectabile de clorură de sodiu 0,9% BP m/v şi glucoză 5% nu este influenţată de prezenţa fosfatului sodic de hidrocortizon. De asemenea, s-a constatat că cefuroxima sodică este compatibilă timp de 24 ore la temperatura camerei atunci când este amestecată în perfuzie i.v. cu:
Heparină (10 şi 50 unităţi/ml) în soluţie de clorură de sodiu pentru preparate injectabile 0,9%; clorură de potasiu (10 şi 40 mEqL) în soluţie de clorură de sodiu pentru preparate injectabile 0,9%.
7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ [A se completa la nivel naţional] {Nume şi adresă} <{tel}> <{fax}> <{e-mail}> 8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ [A se completa la nivel naţional] 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI [A se completa la nivel naţional] 10. DATA REVIZUIRII TEXTULUI [A se completa la nivel naţional]
33

ETICHETAREA
34

INFORMAŢII CARE TREBUIE SĂ APARĂ PE AMBALAJUL SECUNDAR CUTIE 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 500 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 1 g pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 2 g pulbere pentru soluţie perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie perfuzabilă (prezentare Monovial) Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie perfuzabilă (prezentare Monovial) [Vezi Anexa I – a se completa la nivel naţional] Cefuroximă 2. DECLARAREA SUBSTANŢEI(LOR) ACTIVE [A se completa la nivel naţional] 3. LISTA EXCIPIENŢILOR [A se completa la nivel naţional] 4. FORMA FARMACEUTICĂ ŞI CONŢINUTUL [A se completa la nivel naţional] 5. MODUL ŞI CALEA(CĂILE) DE ADMINISTRARE A se citi prospectul înainte de utilizare. Zinacef 250 mg, 750 mg si 1,5 g pulbere pentru soluţie injectabilă; Zinacef 250 mg, 750 mg şi 1,5 g pulbere pentru soluţie injectabilă sau perfuzabilă Administrare intramusculară sau intravenoasă. Zinacef 250 mg, 500 mg, 750 mg şi 1 g pulbere şi solvent pentru soluţie injectabilă Administrare intramusculară sau intravenoasă.
35

Zinacef 750 mg, 1,5 g şi 2 g g pulbere pentru soluţie perfuzabilă Administrare intravenoasă. Zinacef 750 mg şi 1,5 g pulbere pentru soluţie perfuzabilă (prezentare Monovial) Administrare intravenoasă. 6. ATENŢIONARE SPECIALĂ PRIVIND FAPTUL CĂ MEDICAMENTUL NU TREBUIE
PĂSTRAT LA VEDEREA SI ÎNDEMÂNA COPIILOR A nu se lăsa la vederea şi îndemâna copiilor. 7. ALTĂ(E) ATENŢIONARE(ĂRI) SPECIALĂ(E), DACĂ ESTE(SUNT) NECESARĂ(E) 8. DATA DE EXPIRARE EXP 9. CONDIŢII SPECIALE DE PĂSTRARE [A se completa la nivel naţional] 10. PRECAUŢII SPECIALE PRIVIND ELIMINAREA MEDICAMENTELOR NEUTILIZATE SAU A MATERIALELOR REZIDUALE PROVENITE DIN ASTFEL DE MEDICAMENTE, DACĂ ESTE CAZUL 11. NUMELE ŞI ADRESA DEŢINĂTORULUI AUTORIZAŢIEI DE PUNERE PE PIAŢĂ [Vezi Anexa I – a se completa la nivel naţional] { Numele şi adresa} <{tel}> <{fax}> <{e-mail}> 12. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ [A se completa la nivel naţional] 13. SERIA DE FABRICAŢIE Lot 14. CLASIFICARE GENERALĂ PRIVIND MODUL DE ELIBERARE [A se completa la nivel naţional]
36

15. INSTRUCŢIUNI DE UTILIZARE [A se completa la nivel naţional] 16. INFORMAŢII ÎN BRAILLE [A se completa la nivel naţional]
37

MINIMUM DE INFORMAŢII CARE TREBUIE SĂ APARĂ PE AMBALAJELE PRIMARE MICI ETICHETA FLACONULUI 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI ŞI CALEA(CĂILE) DE ADMINISTRARE Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 500 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 1 g pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 2 g pulbere pentru soluţie perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie perfuzabilă (prezentare Monovial) Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie perfuzabilă (prezentare Monovial) Cefuroximă Zinacef 250 mg , 750 mg şi 1,5 g pulbere pentru soluţie injectabilă; Zinacef 250 mg, 750 mg şi 1,5 g pulbere pentru soluţie injectabilă sau perfuzabilă i.m./i.v. Zinacef 250 mg, 500 mg, 750 mg şi 1 g pulbere şi solvent pentru soluţie injectabilă i.m./i.v. Zinacef 750 mg, 1,5 g şi 2 g pulbere pentru soluţie perfuzabilă i.v. Zinacef 750 mg şi 1,5 g pulbere pentru soluţie perfuzabilă (prezentare Monovial) i.v. 2. MODUL DE ADMINISTRARE A se citi prospectul înainte de utilizare. 3. DATA DE EXPIRARE EXP 4. SERIA DE FABRICAŢIE Lot
38
5. CONŢINUTUL PE MASĂ, VOLUM SAU UNITATEA DE DOZĂ [A se completa la nivel naţional] 6. ALTE INFORMAŢII
39

PROSPECTUL
40

PROSPECT: INFORMAŢII PENTRU UTILIZATOR
Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie injectabilă
Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 500 mg pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere şi solvent pentru soluţie injectabilă
Zinacef şi denumirile asociate (vezi Anexa I) 1 g pulbere şi solvent pentru soluţie injectabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie perfuzabilă
Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 2 g pulbere pentru soluţie perfuzabilă
Zinacef şi denumirile asociate (vezi Anexa I) 250 mg pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie injectabilă sau perfuzabilă
Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie injectabilă sau perfuzabilă Zinacef şi denumirile asociate (vezi Anexa I) 750 mg pulbere pentru soluţie perfuzabilă (prezentare
Monovial) Zinacef şi denumirile asociate (vezi Anexa I) 1,5 g pulbere pentru soluţie perfuzabilă(prezentare
Monovial)
Cefuroximă
Citiţi cu atenţie şi în întregime acest prospect înainte de a începe să utilizaţi acest medicament deoarece conţine informaţii importante pentru dumneavoastră.
- Păstraţi acest prospect. S-ar putea să fie necesar să-l recitiţi. - Dacă aveţi orice întrebări suplimentare, adresaţi-vă medicului dumneavoastră sau asistentei medicale. - Dacă manifestaţi orice reacţie adversă, discutaţi cu medicul dumneavoastră sau cu asistenta medicală.
Acestea includ orice posibile reacţii adverse nemenţionate în acest prospect.
Ce găsiţi în acest prospect: 1. Ce este Zinacef şi pentru ce se utilizează 2. Ce trebuie ştiţi înainte să vi se administreze Zinacef 3. Cum se utilizează Zinacef 4. Reacţii adverse posibile 5. Cum se păstrează Zinacef 6. Conţinutul ambalajului şi alte informaţii 1. Ce este Zinacef şi pentru ce se utilizează
Zinacef este un antibiotic utilizat la adulţi şi copii. Acţionează prin distrugerea bacteriilor care determină apariţia infecţiilor. Aparţine unui grup de medicamente denumite cefalosporine.
Zinacef este utilizat pentru tratamentul infecţiilor de la nivelul:
plămânilor sau toracelui tractului urinar pielii şi ţesuturilor moi abdomenului
Zinacef este utilizat, de asemenea, pentru:
a preveni infecţiile în timpul intervenţiilor chirurgicale.
41

2. Ce trebuie să ştiţi înainte să vi se administreze Zinacef Nu trebuie să vi se administreze Zinacef: dacă sunteţi alergic (hipersensibil) la oricare dintre antibioticele din clasa cefalosporinelor sau la
oricare dintre celelalte componente ale Zinacef . Dacă aţi avut vreodată o reacţie alergică severă (hipersensibilitate) la orice alt tip de antibiotic
betalactamic (peniciline, monobactami şi carbapeneme).
Adresaţi-vă medicului dumneavoastră înainte de a începe administrarea Zinacef în cazul în care consideraţi că acestea sunt valabile pentru dumneavoastră. Nu trebuie să vi se administreze Zinacef.
Aveţi grijă deosebită când utilizaţi Zinacef Trebuie să fiţi atenţi la apariţia anumitor simptome cum sunt reacţii alergice şi tulburări gastrointestinale, cum este diareea, pe durata tratamentului cu Zinacef. Acest lucru va reduce riscul de apariţie a unor posibile probleme. Vezi („Afecţiuni cărora este indicat să le acordaţi o atenţie deosebită”) la punctul 4. Dacă aţi avut orice fel de reacţie alergică la alte antibiotice cum este penicilina, este posibil să fiţi alergic şi la Zinacef. Dacă este necesar să vi se efectueze o analiză de sânge sau de urină Zinacef poate influenţa rezultatele analizelor de urină sau de sânge pentru determinarea concentraţiei de glucoză (zahăr) şi ale unei analize denumite testul Coombs. Dacă vi se efectuează aceste analize: Spuneţi persoanei care vă recoltează proba că vi se administrează Zinacef. Alte medicamente şi Zinacef Spuneţi medicului dumneavoastră dacă luaţi, aţi început recent sau dacă începeţi să luaţi orice alte medicamente. Acestea includ medicamentele pe care le puteţi lua fără prescripţie medicală. Unele medicamente pot influenţa acţiunea Zinacef sau pot creşte probabilitatea să prezentaţi reacţii adverse. Acestea includ:
antibiotice aminoglicozidice medicamente pentru eliminarea excesului de apă (diuretice), cum este furosemid probenecid anticoagulante orale Spuneţi medicului dumneavoastră dacă această situaţie este valabilă în cazul dumneavoastră. Este
posibil să aveţi nevoie de controale suplimentare, pentru a vi se monitoriza funcţia rinichilor pe durata terapiei cu Zinacef.
Contraceptive orale Zinacef poate reduce eficacitatea contraceptivelor orale. Dacă luaţi contraceptive orale în timp ce vi se administrează Zinacef trebuie să utilizaţi şi o metodă de contracepţie de tip barieră (cum sunt prezervativele). Cereţi sfatul medicului dumneavoastră. Sarcina, alăptarea şi fertilitatea Spuneţi medicului înainte de a vi se administra Zinacef: dacă sunteţi gravidă, credeţi că aţi putea fi gravidă sau intenţionaţi să rămâneţi gravidă dacă alăptaţi Medicul dumneavoastră va lua în considerare beneficiul administrării tratamentului cu Zinacef pentru dumneavoastră şi riscul pentru copil. Conducerea vehiculelor şi folosirea utilajelor Nu conduceţi vehicule sau nu folosiţi utilaje dacă nu vă simţiţi bine. Informaţii importante privind unele componente ale Zinacef Zinacef conţine sodiu. Trebuie să ţineţi cont de acest aspect în cazul în care urmaţi o dietă cu conţinut controlat de sodiu.
42

Concentraţia Zinacef Cantitate per flacon
250 mg 14 mg 500 mg 28 mg 750 mg 42 mg
1 g 56 mg 1,5 g 83 mg 2 g 111 mg
3. Cum să utilizaţi Zinacef Zinacef este administrat de obicei de către un medic sau de către o asistentă. Poate fi administrat sub formă de picătură cu picătură (perfuzie intravenoasă) sau injectabil direct în venă sau în muşchi. Doza uzuală Medicul dumneavoastră va decide doza corectă de Zinacef care vă va fi administrată, aceasta depinzând de: severitatea şi tipul infecţiei, dacă sunteţi tratat cu alte antibiotice, greutatea şi vârsta dumneavoastră, cât de bine vă funcţionează rinichii. Nou-născuţi (0-3 săptămâni) Pentru fiecare kg de greutate corporală, li se va administra între 30 mg şi 100 mg Zinacef pe zi, fracţionat în două sau trei doze . Sugari (peste trei săptămâni) şi copii Pentru fiecare kg de greutate corporală, li se va administra între 30 mg şi 100 mg Zinacef pe zi, fracţionat în trei sau patru doze. Adulţi şi adolescenţi Între 750 mg şi 1,5 g Zinacef pe zi, fracţionat în două, trei sau patru doze. Doza maximă: 6 g pe zi. Pacienţi cu probleme ale rinichilor În cazul în care aveţi o problemă a rinichilor, medicul dumneavoastră vă poate modifica doza. Discutaţi cu medicul dumneavoastră dacă această situaţie este valabilă în cazul dumneavoastră. 4. Reacţii adverse posibile Ca toate medicamentele, Zinacef poate provoca reacţii adverse, cu toate că nu apar la toate persoanele. Afecţiuni cărora este indicat să le acordaţi o atenţie deosebită Un număr mic de persoane tratate cu Zinacef prezintă o reacţie alergică sau o reacţie la nivelul pielii potenţial gravă. Simptomele acestor reacţii includ: reacţie alergică severă. Semnele includ erupţie în relief pe piele însoţită de mâncărimi, umflare,
uneori la nivelul feţei sau gurii, care determină dificultăţi la respiraţie.
Erupţii tecătoare pe piele, care pot forma vezicule, cu aspect de mici ţinte (un punct central întunecat înconjurat de o zonă mai deschisă şi cu margini circulare închise la culoare).
erupţie pe piele, extinsă, cu vezicule şi exfoliere a pielii. (Acestea pot fi semne ale sindromului Stevens-Johnson sau ale necrolizei epidermice toxice).
infecţiile fungice apar în cazuri rare, medicamentele ca Zinacef pot determina creşterea excesivă a ciupercilor (Candida) în organism, fapt care poate cauza infecţiile fungice (cum este candidoza orală).
43

Probabilitatea de apariţie a acestei reacţii adverse este mai mare în cazul în care sunteţi tratat cu Zinacef o perioadă lungă de timp.
Adresaţi-vă imediat medicului sau asistentei în cazul în care aveţi oricare dintre aceste simptome.
Reacţii adverse frecvente
Acestea pot afecta până la 1 din 10 persoane:
durere la locul injectării, umflare şi înroşire de-a lungul venei.
Spuneţi medicului dumneavoastră dacă vă deranjează oricare dintre acestea.
Reacţii adverse frecvente care pot să apară la analizele de sânge:
creşteri ale concentraţiilor unor substanţe (enzime) produse de ficat modificări ale numărului de celule albe din sânge (neutropenie sau eozinofilie) număr redus de celule roşii din sânge (anemie)
Reacţii adverse mai puţin frecvente
Acestea pot afecta până la 1 din 100 de persoane: erupţie trecătoare pe piele, însoţită de mâncărimi, în relief (urticarie) diaree, greaţă, durere de stomac Spuneţi medicului dumneavoastră în cazul în care aveţi oricare dintre acestea. Reacţii adverse mai puţin frecvente care pot să apară la analizele de sânge:
număr redus de celule albe din sânge (leucopenie) creştere a concentraţiei bilirubinei (o substanţă produsă de ficat)
rezultate pozitive la testul Coombs.
Alte reacţii adverse Alte reacţii adverse au apărut la un număr foarte mic de persoane, însă frecvenţa acestora nu este cunoscută cu precizie:
infecţii fungice temperatură mare (febră) reacţii alergice inflamaţie a colonului (intestinului gros), care determină diaree, de obicei cu eliminare de sânge şi
mucus, durere de stomac inflamaţie a rinichilor şi vaselor de sânge
distrugerea prea rapidă a celulelor roşii din sânge (anemie hemolitică).
erupţie pe piele, care poate determina apariţia unor vezicule cu aspect de mici ţinte (un punct central închis la culoare înconjurat de o zonă mai deschisă şi cu margini circulare închise la culoare), eritem polimorf.
Spuneţi medicului dumneavoastră în cazul în care aveţi oricare dintre acestea. Reacţii adverse care pot să apară la analizele de sânge:
reducere a numărului de trombocite din sânge (celule care ajută la coagularea sângelui - trombocitopenie)
creştere a concentraţiilor de azot ureic şi de creatinină în sânge.
44

În cazul în care aveţi reacţii adverse Spuneţi medicului dumneavoastră sau farmacistului. Acestea includ orice posibile reacţii adverse
nemenţionate în acest prospect. 5. Cum se păstrează Zinacef [A se completa la nivel naţional] Nu lăsaţi acest medicament la vederea şi îndemâna copiilor. Nu utilizaţi acest medicament după data de expirare înscrisă pe cutie, după EXP. Data de expirare se referă la ultima zi a lunii respective. Nu aruncaţi niciun medicament pe calea apei sau a reziduurilor menajere. Întrebaţi farmacistul cum să aruncaţi medicamentele pe care nu le mai folosiţi. Aceste măsuri vor ajuta la protejarea mediului. 6. Conţinutul ambalajului şi alte informaţii
Ce conţine Zinacef
[A se completa la nivel naţional]
Cum arată Zinacef şi conţinutul ambalajului [A se completa la nivel naţional] Deţinătorul autorizaţiei de punere pe piaţă şi fabricantul [A se completa la nivel naţional] Acest medicament este autorizat în Statele Membre ale Spaţiului Economic European sub următoarele denumiri comerciale: 250 mg pulbere pentru soluţie injectabilă Austria – Curocef Danemarca, Finlanda, Grecia, Ungaria, Irlanda, Lituania, Malta, Olanda, Norvegia, Polonia, Suedia, Marea Britanie – Zinacef Italia– Curoxim Franţa – Zinnat 500 mg pulbere pentru soluţie injectabilă Italia– Curoxim 750 mg pulbere pentru soluţie injectabilă sau perfuzabilă Austria – Curocef Belgium, Bulgaria, Cipru, Republica Cehă, Danemarca, Estonia, Finlanda, Grecia, Ungaria, Islanda, Irlanda, Lituania, Luxemburg, Malta, Olanda, Norvegia, Polonia, Portugalia, România, Slovenia, Suedia, Marea Britanie – Zinacef Italia– Curoxim Franţa - Zinnat 1 g pulbere pentru soluţie injectabilă sau perfuzabilă Italia– Curoxim
45

1,5 g pulbere pentru soluţie injectabilă sau perfuzabilă Austria – Curocef Belgium, Bulgaria, Cipru, Republica Cehă, Danemarca , Estonia, Finlanda, Grecia, Ungaria, Islanda, Irlanda, Lituania, Luxemburg, Olanda, Norvegia, Polonia, România, Slovenia, Suedia, Marea Britanie – Zinacef Franţa – Zinnat 2 g pulbere pentru soluţie perfuzabilă Italia– Curoxim 750 mg Monovial pulbere pentru soluţie perfuzabilă Italia– Curoxim 1,5 g flacon Monovial pulbere pentru soluţie perfuzabilă Italia– Curoxim l Acest prospect a fost aprobat în { LL/AAAA }. <----------------------------------------------------------------------------------------------------------------------------- Următoarele informaţii sunt destinate numai medicilor şi profesioniştilor din domeniul sănătăţii: Instrucţiuni de reconstituire Volumul care trebuie adăugat şi concentraţiile soluţiei obţinute, de utilizat în cazul în care este necesară administrarea de doze fracţionate.
Volumul care trebuie adăugat şi concentraţiile soluţiei obţinute, de utilizat în cazul în care este necesară administrarea de doze fracţionate
Volumul flaconului Cantitatea de apă care trebuie adăugată (ml)
Concentraţia aproximativă a
cefuroximei (mg/ml)**
250 mg pulbere pentru soluţie injectabilă 250 mg intramuscular
intravenos 1 ml până la minimum 2 ml
216 116
500 mg pulbere pentru soluţie injectabilă 500 mg intramuscular
2 ml 216
750 mg pulbere pentru soluţie injectabilă sau perfuzabilă 750 mg intramuscular
intravenos în bolus perfuzie intravenoasă
3 ml până la minimum 6 ml până la minimum 6 ml
216 116 116
1 g pulbere pentru soluţie injectabilă 1 g
intramuscular intravenos în bolus
4 ml 10 ml
216 94
1,5 g pulbere pentru soluţie injectabilă sau perfuzabilă 1,5 g
intramuscular intravenos în bolus perfuzie intravenoasă
6 ml până la minimum15ml 15 ml*
216 94 94
2 g pulbere pentru soluţie perfuzabilă 2 g
Perfuzie intravenoasă 20 ml 94
46

47
* Soluţia reconstituită care se va adăuga la 50 sau 100 ml de soluţie perfuzabilă compatibilă (vezi informaţiile referitoare la compatibilitate, mai jos) ** Volumul soluţiei de cefuroximă rezulat în mediul de reconstituire este crescut din cauza factorului de deplasare a substanţei, rezultat din concentraţiile listate în mg/ml. Zinacef 750 mg şi 1,5 g pulbere pentru soluţie perfuzabilă (prezentare Monovial) Pregătirea soluţiei pentru perfuzie intravenoasă Conţinutul flaconului Monovial se adaugă în pungi pentru perfuzie cu volum mic care conţin soluţie de clorură de sodiu 0,9% pentru preparate injectabile sau soluţie de glucoză 5% pentru preparate injectabile sau alte soluţii compatibile.
1. Îndepărtaţi partea superioară detaşabilă a etichetei şi scoateţi capacul. 2. Introduceţi acul flaconului Monovial în dispozitivul de conectare al pungii pentru perfuzie. 3. Pentru activare, împingeţi suportul din plastic al acului flaconului Monovial spre gulerul flaconului,
până se aude un „clic”. 4. Ţinându-l în poziţie verticală, umpleţi flaconul până la aproximativ două treimi din capacitate,
strângând de câteva ori punga. 5. Agitaţi flaconul pentru a reconstitui soluţia care conţine cefuroximă sodică. 6. Ţinând flaconul deasupra celorlalte componente, transferaţi soluţia reconstituită care conţine
cefuroximă sodică în punga de perfuzie, strângând şi slăbind alternativ punga. 7. Repetaţi etapele 4 – 6, pentru a clăti interiorul flaconului. Eliminaţi în condiţii de siguranţă flaconul
gol de Monovial.Verificaţi dacă pulberea s-a dizolvat şi dacă punga nu prezintă scurgeri. Compatibilitate Cefuroximă sodică 1,5 g reconstituită cu 15 ml apă pentru preparate injectabile poate fi adăugată soluţiei injectabile care conţine metronidazol (500 mg/100 ml) şi ambele îşi menţin activitatea timp de până la 24 ore, la temperaturi sub 25°C. Cefuroximă sodică 1,5 g este compatibilă cu azlocilină 1 g (în 15 ml) sau 5 g (în 50 ml) timp de până la 24 ore, la temperatura de 4°C sau timp de 6 ore la temperaturi sub 25°C. Cefuroxima sodică (5 mg/ml) în soluţie injectabilă cu xilitol 5% m/v sau 10% m/v poate fi păstrată timp de până la 24 ore la 25°C. Cefuroxima sodică este compatibilă cu soluţia care conţine clorhidrat de lidocaină până la 1%. Cefuroxima sodică este compatibilă cu următoarele soluţii perfuzabile. Îşi va menţine potenţa timp de până la 24 ore la temperatura camerei în:
Clorură de sodiu 0,9% m/v soluţie pentru preparate injectabile BP Glucoză 5% soluţie pentru preparate injectabile BP Clorură de sodiu 0,18% m/v plus glucoză 4% soluţie pentru preparate injectabile BP Glucoză 5% şi clorură de sodiu 0,9% soluţie pentru preparate injectabile Glucoză 5% şi clorură de sodiu 0,45% soluţie pentru preparate injectabile Glucoză 5% şi clorură de sodiu 0,225% soluţie pentru preparate injectabile Glucoză 10% soluţie pentru preparate injectabile Zahăr invertit 10% în apă pentru preparate injectabile Soluţie injectabilă Ringer USP Soluţie injectabilă Ringer lactat USP Soluţie injectabilă de lactat de sodiu M/6 Soluţie injectabilă de lactat de sodiu compus BP (soluţie Hartmann).
Stabilitatea cefuroximei sodice în soluţie pentru preparate injectabile de clorură de sodiu 0,9% BP m/v şi glucoză 5% nu este influenţată de prezenţa fosfatului sodic de hidrocortizon. De asemenea, s-a constatat că cefuroxima sodică este compatibilă timp de 24 ore la temperatura camerei atunci când este amestecată în perfuzie i.v. cu:
Heparină (10 şi 50 unităţi/ml) în soluţie de clorură de sodiu pentru preparate injectabile 0,9%; clorură de potasiu (10 şi 40 mEqL) în soluţie de clorură de sodiu pentru preparate injectabile 0,9%.







