Page 1
1
Universitatea “Alexandru Ioan Cuza” Iași
Facultatea de Fizică
REZUMATUL TEZEI DE DOCTORAT
CONTRIBUŢII LA STUDIUL INTERACŢIUNILOR
INTERMOLECULARE ÎN MEDII CONDENSATE
Conducător ştiinţific,
Prof. univ.em. dr. Dana Ortansa Dorohoi
Doctorand,
Cezarina Călugăru (Moroșanu)
Iaşi-2018
Page 2
2
Mulțumiri
Mulțumesc doamnei prof. univ. em. dr. Dana Ortansa Dorohoi, conducătoarea
tezei de doctorat, pentru încrederea acordată, pentru sprijin, pentru îndrumare cu
competență și profesionalism pe toată perioada școlii doctorale.
Mulțumesc membrilor Comisiei de îndrumare a tezei, respectiv doamnei prof.
univ. dr. Dorina Emilia Creangă, domnului conf. univ. dr. Dan Gheorghe Dimitriu și
domnului conf. univ. dr. Silviu Gurlui pentru recomandările făcute atât pe perioada
cercetării, cât și în realizarea tezei.
Mulțumesc conducerii Facultății de Fizică din Iași, domnului decan - conf. univ.
dr. Sebastian Popescu, pentru sprijinul logistic și financiar, necesar activității de cercetare.
De asemenea, mulțumirile mele se îndreaptă și către membrii Comisiei de
examinare a tezei: prof. univ. dr. Diana Mardare – președintele comisiei, prof. univ. dr.
Simion Aștilean, cercet. pr. dr. Maria Bercea și conf. univ. dr. Dan Gheorghe Dimitriu –
referenți științifici, care mi-au analizat teza și mi-au oferit sugestii referitoare la diferite
aspecte ale lucrării.
Mulțumesc familiei și prietenilor pentru sprijinul, încrederea și încurajarea
oferite, foarte necesare în toată această perioadă.
Page 3
3
UNIVERSITATEA “ALEXANDRU IOAN CUZA”, IAȘI
Școala Doctorală de Fizică
Vă fac cunoscut faptul că în data de 20 septembrie 2018, ora 11:00, în Sala L1, doamna Cezarina
CĂLUGĂRU (căs. MOROȘANU) va susține, în ședință publică, teza de doctorat:
“CONTRIBUȚII LA STUDIUL INTERACȚIUNILOR INTERMOLECULARE ÎN MEDII
CONDENSATE”
în vederea obținerii titlului științific de doctor în domeniul Științe Exacte – Fizică, subdomeniul
Optică și spectroscopie.
Comisia de examinare a tezei:
Prof. univ. dr. Diana MARDARE
Președinte
Directorul Școlii Doctorale de Fizică
Universitatea “Alexandru Ioan Cuza”, Iași
Prof. univ. em. dr. Dana Ortansa DOROHOI
Conducător științific
Facultatea de Fizică
Universitatea “Alexandru Ioan Cuza”, Iași
Prof. univ. dr. Simion AȘTILEAN
Referent
Facultatea de Fizică
Universitatea “Babeș Bolyai”, Cluj-Napoca
CP 1 dr. Maria BERCEA
Referent
Institutul de Chimie Macromoleculară “Petru Poni”, Iași
Conf. univ. dr. Dan Gheorghe DIMITRIU
Referent
Facultatea de Fizică
Universitatea “Alexandru Ioan Cuza”, Iași
Vă invităm, pe această cale, să participați la ședința publică de susținere a tezei.
Page 4
4
Cuprins
Obiective inițiale ................................................................ ................................................................. 7
Introducere .......................................................................................................................................... 8
Partea I. Interacțiuni intermoleculare în medii condensate – studiu din literatură ............................ 11
Capitolul I. Interacțiuni intermoleculare în lichide ........................................................................... 11
I.1. Forțe intermoleculare în lichide ..................................................................... ............................. 11
I.2. Modele de lichide ................................................................................... ..................................... 11
I.2.1. Modelul statistic al soluțiilor cu trei componente .................................................................... 12
I.2.2. Manifestări ale interacțiunilor de dispersie din starea lichidă în spectrele electronice de
absorbție ............................................................................................ ................................................ 14
Partea a II-a. Contribuții personale ................................................................ ................................... 17
Capitolul II. Caracterizarea cuanto-mecanică și solvatocromică a quercetinei ................................ 17
II.1. Introducere ........................................................................................................ ......................... 17
II.2. Detalii computaționale și experimentale ................................................... ................................ 18
II.3. Rezultate și discuții .......................................................................................... .......................... 19
II.3.1. Caracterizare cuanto-mecanică a quercetinei ......................................................................... 19
II.3.2. Rezultate spectrale ......................................................... ......................................................... 23
II.4. Concluzii .................................................................................................................................... 27
Capitolul III. Studiul computațional și solvatocromic al moleculelor de piridiniu-acetil-benzoil-
metilid .............................................................................. ................................................................. 27
III.1. Introducere ............................................................................................................................... 27
III.2. Detalii computaționale și experimentale .................................................................................. 28
III.3. Rezultate și discuții .................................................................................................................. 30
Page 5
5
III.3.1. Rezultate computaționale ...................................................................................................... 30
III.3.2. Rezultate spectrale ................................................................................ ................................ 32
III.3.2.1. Soluții binare ...................................................................................................................... 32
III.3.2.2. Soluții ternare ....................................................................... .............................................. 35
III.4. Concluzii .................................................................................................................................. 37
Capitolul IV. Momentul de dipol al moleculei de albastru de timol în stare excitată, estimat prin
studii solvatocromice și de mecanică cuantică .................................................................................. 38
IV.1. Introducere ................................................................. .............................................................. 38
IV.2. Detalii experimentale și computaționale .................................................................................. 39
IV.3. Rezultate și discuții .................................................................................................................. 39
IV.3.1. Rezultate computaționale ...................................................................................................... 39
IV.3.2. Rezultate spectrale ....................................................... ......................................................... 41
IV.4. Concluzii ........................................................................................... ....................................... 44
Capitolul V. Studiul cuanto-mecanic și spectral al fluoresceinei ..................................................... 45
V.1. Introducere .................................................................................................... ............................ 45
V.2. Detalii computaționale și experimentale ................................................................................... 45
V.3. Rezultate și discuții ................................................................................................................... 46
V.3.1. Caracterizarea cuanto-mecanică a fluoresceinei .................................................................... 46
V.3.2. Rezultate spectrale ................................................................. ................................................. 49
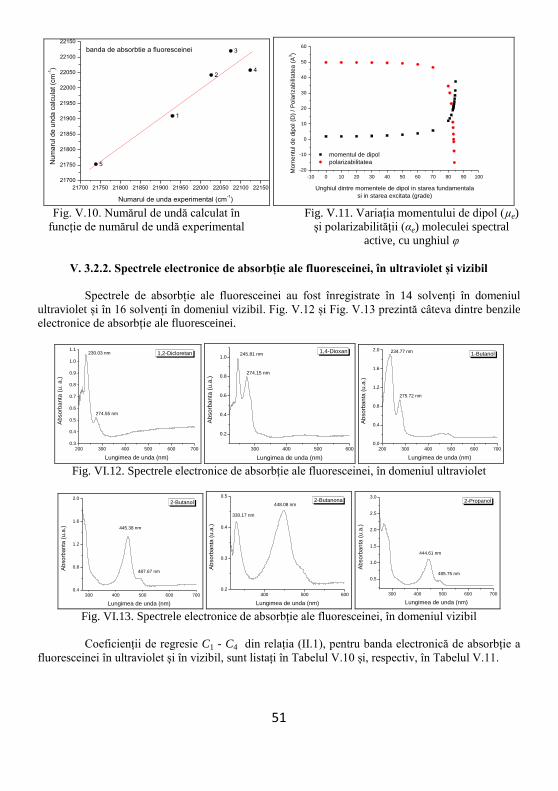
V.3.2.1. Spectrele electronice de absorbție ale fluoresceinei în solvenți hidroxilici ........................ 49
V.3.2.2. Spectrele electronice de absorbție ale fluoresceinei, în ultraviolet și vizibil ...................... 51
V.3.3. Estimarea momentului de dipol al fluoresceinei în starea excitată ........................................ 53
V.4. Concluzii ................................................................................................... ................................ 54
Concluzii generale ............................................................................................ ................................. 54
Page 6
6
Perspective .................................................................................................................... .................... 56
Bibliografie ........................................................................................ ............................................... 56
Anexe .......................................................................... .................................................................... 62
Obs:
Rezumatul tezei de doctorat păstrează numerotarea capitolelor, subcapitolelor, figurilor,
formulelor și a tabelelor din teză.
Page 7
7
Obiective inițiale
1) Optimizarea structurii geometrice a moleculelor studiate cu ajutorul unor programe de
modelare moleculară și determinarea unor parametri moleculari corespunzători stării
electronice fundamentale.
2) Studiul cuanto-mecanic și spectral al unor compuși organici.
3) Îmbinarea rezultatelor din studiul cuanto-mecanic cu cele din studiul solvatocromic, în
vederea obținerii unor informații despre starea excitată a moleculelor.
4) Stabilirea naturii interacțiunilor moleculare în soluțiile moleculelor spectral active și
estimarea procentuală a contribuției fiecărui tip de interacțiune la deplasarea spectrală
totală a benzilor electronice de absorbție sub influența solventului.
5) Determinarea, printr-o metodă variațională, a momentului de dipol în starea excitată a
moleculelor spectral active.
6) Determinarea unghiului dintre momentele de dipol în starea fundamentală și în starea
excitată a moleculelor studiate.
7) Utilizarea unor soluții ternare în vederea determinării diferenței dintre energia de
interacțiune în perechile de molecule.
8) Estimarea tăriei interacțiunilor specifice prin alegerea corespunzătoare a solvenților
utilizați (unul activ și unul inactiv din punctul de vedere al interacțiunilor specifice).
Page 8
8
Introducere
Studiul substanțelor în stare condensată, în special studiul lichidelor, se face prin metode
experimentale, utilizând spectrele moleculare de absorbție. Se utilizează o substanță spectral activă,
introdusă în concentrație foarte mică în lichidul pur. Concentrația substanței spectral active fiind
foarte mică, distanțele dintre moleculele spectral active sunt relativ mari și interacțiunile dintre
aceste molecule sunt neglijabile. Prin urmare, soluția poate fi considerată ca fiind alcătuită din
subsisteme, fiecare subsistem fiind format dintr-o moleculă spectral activă, înconjurată de molecule
de lichid. În starea de gaz ideal (vapori), se consideră că aceste molecule nu interacționează între
ele, forțele de interacțiune fiind neglijabile. La trecerea din starea de gaz ideal în stare lichidă,
forțele intermoleculare cresc semnificativ și aceste interacțiuni dintre moleculele solutului și
moleculele solventului determină modificări ale spectrelor electronice. Solventul utilizat trebuie să
fie transparent în domeniul în care se înregistrează spectrul astfel încât, în starea excitată a
sistemului, numai moleculele spectral active (moleculele solutului) sunt în stare excitată. Din aceste
deplasări spectrale se pot obține informații despre parametrii electro-optici ai moleculelor spectral
active în starea excitată (moment de dipol, polarizabilitate), atunci când se cunosc sau se determină
valorile acestor parametri în stare fundamentală prin alte mijloace (de exemplu, prin calcule cuanto-
mecanice).
Partea I a tezei conține aspecte teoretice din literatura de specialitate, care au stat la baza
cercetărilor ulterioare. În Capitolul I este prezentată o clasificare a forțelor intermoleculare în
lichide. De asemenea, este descris modelul statistic al soluțiilor cu trei componente. În cadrul acestui
model, se utilizează un amestec de doi solvenți transparenți în domeniul în care se înregistrează
spectrul, în care se introduce o substanță spectral activă, în concentrație foarte mică. Soluția ternară
poate fi reprezentată printr-un subsistem ce conține o moleculă spectral activă și moleculele celor
doi solvenți, aflate în primul strat de solvatare. Modelul a fost utilizat în această lucrare pentru a
determina energia de interacțiune în perechile de molecule de tipul moleculă spectral activă –
moleculă solvent activ, respectiv moleculă spectral activă – moleculă solvent inactiv (din punctul de
vedere al interacțiunilor intermoleculare). În Capitolul I este analizată și influența interacțiunilor de
dispersie (care se manifestă în lichide cu molecule neutre și nepolare) asupra deplasărilor din
spectrele electronice de absorbție.
Partea a II-a a tezei conține contribuții personale la studiul interacțiunilor intermoleculare
în lichide. Au fost studiate soluțiile binare și ternare ale unor substanțe utilizate în medicină,
farmacie, cercetare și industrie (quercetină, piridiniu-acetil-benzoil-metilid, albastru de timol,
fluoresceină).
În Capitolul II este realizată o caracterizare cuanto-mecanică și solvatocromică a
quercetinei, un flavonoid care se găsește în fructe, legume, frunze, semințe și nuci, având un
potențial foarte mare de utilizare în aplicații terapeutice (efecte vasodilatator, antihipertensiv,
biomaterial pentru regenerarea oaselor, terapia cancerului, efecte împotriva unor virusuri, potențial
tratament împotriva bolii Alzheimer, proprietăți anti-îmbătrânire și de întinerire).
Structura geometrică a moleculei de quercetină a fost optimizată cu ajutorul programului
de modelare moleculară Spartan’14, utilizând metoda funcționalei de densitate. Prin calcule cuanto-
mecanice, programul Spartan furnizează valorile unor parametri moleculari (energii, moment de
dipol și polarizabilitate în starea fundamentală a moleculei, volum, arie, coeficient de partiție),
precum și reprezentări grafice ale unor mărimi fizice care reprezintă indicatori ai adiției
electrofile/nucleofile și care dau informații despre reactivitatea chimică a moleculei (harta
potențialului electrostatic, harta potențialului de ionizare locală și harta |LUMO|).
Numerelor de undă din maximele benzilor electronice de absorbție ale quercetinei li s-a
aplicat o metodă de regresie liniară multiplă care a permis analiza modului în care parametrii
Page 9
9
macroscopici și parametrii Kamlet-Taft ai solvenților influențează deplasările spectrale. A fost
estimată contribuția fiecărui tip de interacțiune la deplasarea spectrală totală (orientare-inducție,
dispersie-polarizare, interacțiuni specifice). S-a obținut o bună corelație liniară între valorile
experimentale ale numerelor de undă și cele calculate, pe baza relației de regresie liniară multiplă.
De asemenea, a fost studiată dependența momentului de dipol și polarizabilității moleculei de
quercetină în starea excitată, de unghiul dintre momentele de dipol în stările fundamentală și
excitată.
Capitolul III al tezei conține studiul computațional și solvatocromic al moleculelor de
piridiniu-acetil-benzoil-metilid (PABM). Cicloimoniu-ilidele sunt substanțe puternic nucleofile, pot
participa la reacții de cicloadiție și reprezintă o sursă de noi compuși heterociclici. Spectrele de
absorbție ale piridiniu-ilidelor dau informații despre structura moleculară și despre natura și tăria
interacțiunilor intermoleculare în soluțiile de ilidă. Piridiniu-ilidele sunt utilizate ca indicatori acido-
bazici, precursori în reacțiile de obținere a unor noi compuși heterociclici, materiale
semiconductoare în filme subțiri, substanțe antimicrobiene și antifungice.
Au fost estimate momentul de dipol electric și polarizabilitatea moleculei de PABM în
starea excitată, utilizând doar numerele de undă ale benzii de transfer intramolecular de sarcină din
vizibil, în diferiți solvenți. Pentru optimizarea geometriei moleculei, precum și pentru determinarea
unor parametri electro-optici ai moleculei în starea fundamentală, a fost utilizat programul Spartan.
Reprezentând grafic valoarea experimentală a numărului de undă în funcție de constanta
dielectrică a solventului utilizat, s-a constatat separarea, în apropierea a două curbe diferite, a
punctelor corespunzătoare solvenților neprotici și punctelor corespunzătoare solvenților hidroxilici.
Distanța dintre aceste curbe este o măsură a interacțiunilor specifice ale ilidei cu solvenții
hidroxilici. În limitele modelului statistic al soluțiilor ternare, s-a determinat diferența dintre
energiile potențiale în perechile moleculare de tipul ilidă – solvent protic și ilidă – solvent neprotic,
cele două seturi de soluții ternare studiate fiind PABM + octanol (1) + 1,2 dicloretan (2) și,
respectiv, PABM + acid propionic (1) + cloroform (2).
A fost estimată contribuția fiecărui tip de interacțiune la deplasarea spectrală totală.
Utilizând valorile polarizabilității și momentului de dipol în starea fundamentală, precum și valorile
coeficienților care descriu contribuția interacțiunilor universale la deplasările spectrale totale, s-a
studiat variația momentului de dipol și polarizabilității în starea excitată a moleculei PABM, în
funcție de unghiul dintre momentele de dipol în stare fundamentală și în stare excitată.
Capitolul IV este dedicat albastrului de timol, o substanță cu foarte multe aplicații în
diferite domenii (studiul schimbului de protoni în reacțiile organice, diferite tipuri de senzori,
dozimetre, reactiv color în detectarea proteinelor, construcția dispozitivelor de afișare și a celulelor
de combustibil, fabricarea de produse cosmetice, indicatori pentru determinarea creșterii bacteriilor
etc.).
A fost realizat un studiu spectral și cuanto-chimic al albastrului de timol, pentru a stabili
parametrii moleculari ai acestei substanțe în stările electronice responsabile pentru apariția benzii de
absorbție din vizibil, parametri care sunt indicatori ai reactivității sale și ai abilității de a penetra
membranele celulare.
S-a obținut o dependență liniară foarte bună între valorile experimentale și valorile
calculate ale numerelor de undă, pe baza regresiei liniare multiple, s-a estimat contribuția fiecărui tip
de interacțiune la deplasarea spectrală totală și a fost evaluată variația momentului de dipol electric
al moleculei în timpul procesului de absorbție.
În Capitolul V, substanța studiată din punct de vedere cuanto-mecanic și spectral a fost
fluoresceina, o substanță utilizată în mod frecvent în biologia celulară, medicină, chimie,
optoelectronică, hidrografie și criminologie. Pentru a determina contribuția interacțiunilor specifice
Page 10
10
la deplasarea spectrală totală, au fost efectuate măsurători utilizând un număr mare de solvenți, atât
solvenți hidroxilici cât și solvenți fără gruparea –OH (solvenți neprotici).
Pentru soluțiile obținute doar cu alcooli, se poate considera că interacțiunile specifice
afectează în aceeași manieră poziția benzii de absorbție, astfel încât a fost utilizată o dependență
multiliniară pentru a descrie doar influența interacțiunilor de orientare-inducție și dispersie-
polarizare asupra deplasărilor spectrale ale fluoresceinei în solvenții utilizați.
Proprietățile moleculare ale fluoresceinei au fost calculate utilizând două metode diferite
ale programului Spartan: metoda semi-empirică PM3 și metoda funcționalei de densitate. Modelele
semi-empirice sunt cele mai simple metode bazate pe mecanica cuantică, pot fi aplicate moleculelor
ce conțin 100-200 de atomi și sunt adecvate pentru evaluarea unor proprietăți care depind doar de
geometria moleculei. Metoda funcționalei de densitate necesită resurse de timp mai mari, dar
furnizează rezultate mult mai bune.
Utilizând numerele de undă corespunzătoare maximelor benzilor electronice de absorbție
ale fluoresceinei, în vizibil și ultraviolet, precum și parametrii furnizați de programul Spartan, au
fost evaluate contribuțiile interacțiunilor de orientare-inducție, dispersie-polarizare, precum și
contribuțiile interacțiunilor specifice, la deplasarea spectrală totală în diferiți solvenți. A fost
utilizată dependența coeficienților de regresie liniară care exprimă interacțiunile universale, de
parametrii microscopici ai moleculei de fluoresceină, pentru a estima parametrii electro-optici în
starea fundamentală a moleculei.
Momentele de dipol determinate prin metoda variațională folosind numai spectrele
electronice de absorbție pentru moleculele studiate aflate în stări excitate, au valori concordante cu
cele determinate în literatura de specialitate folosind date spectrale atât pentru absorbție cât și pentru
fluorescență.
Page 11
11
Partea I. Interacțiuni intermoleculare în medii condensate – studiu din literatură
Capitolul I. Interacțiuni intermoleculare în lichide
I.1. Forțe intermoleculare în lichide
Moleculele oricărei substanțe sunt alcătuite din particule încărcate cu sarcini electrice.
Interacțiunile dintre sarcinile electrice ale acestor sisteme atomice determină forțele intermoleculare.
Mărimea forțelor de interacțiune dintre molecule depinde de distanța dintre sistemele atomice,
forțele fiind cu atât mai mici cu cât distanțele sunt mai mari.
Mărimea forțelor intermoleculare depinde și de starea de agregare a substanței. Cele mai
mari valori ale forțelor intermoleculare sunt în substanțele aflate în stare solidă și lichidă (numite
medii condensate), iar cele mai mici valori ale forțelor de interacțiune dintre sistemele atomice se
înregistrează în substanțele în stare de agregare gazoasă. Se disting trei tipuri de forțe
intermoleculare, în funcție de natura lor: de atracție, de repulsie și cu transfer de sarcină.
Forțele de atracție sunt forțe nesaturate, ceea ce înseamnă că o moleculă poate interacționa
simultan cu un număr mare de alte molecule. La rândul lor, forțele de atracție pot fi clasificate în
funcție de cele trei tipuri de interacțiuni care se manifestă între molecule, și anume:
interacțiunile dintre moleculele ce posedă momente permanente de dipol electric (forțe de
orientare);
interacțiunile dintre momentele permanente de dipol electric ale unor molecule și
momentele de dipol induse de acestea în molecule ce nu posedă momente permanente de
dipol (forțe de inducție);
interacțiunile dintre momentele de dipol instantanee ale moleculelor (datorate fluctuațiilor
instantanee din mișcarea electronilor de valență) și momentele induse de acestea în alte
sisteme atomice (forțe de dispersie).
Forțele intermoleculare de repulsie se manifestă atunci când distanța dintre sistemele
atomice scade sub o anumită valoare și predomină respingerea dintre nuclee și respingerea dintre
învelișurile electronice ale atomilor.
Forțele intermoleculare cu transfer de sarcină se manifestă la formarea complecșilor
moleculari și acționează pe distanțe de ordinul diametrelor moleculare.
I.2. Modele de lichide
Pentru investigarea stării lichide au fost elaborate diferite modele, precum modelul cinetic
al unui lichid simplu, modelul celular Abe pentru un lichid pur și modelul statistic al soluțiilor cu
trei componente.
Pentru elaborarea modelelor cinetice pentru starea lichidă s-a plecat de la modelele
existente pentru gazele condensate, având în vedere faptul că, în apropierea punctului critic, unele
proprietăți fizice ale gazului condensat și ale lichidului sunt comune. Modelul cinetic al unui lichid
simplu utilizează următoarele ipoteze: sistemele atomice componente sunt identice, particulele
interacționează în perechi și energia potențială totală a sistemului este suma energiilor de
interacțiune ale perechilor de particule. Acest model permite calculul integralei de configurație a
unui sistem de particule care interacționează prin forțe van der Waals.
Modelul propus de Takehiro Abe se aplică lichidelor cu molecule suficient de mici, pentru
a putea fi considerate de formă sferică, molecule între care se exercită forțe de tip van der Waals.
Acest model permite estimarea unor mărimi microscopice caracteristice moleculelor.
Page 12
12
Modelul statistic al soluțiilor cu trei componente utilizează un amestec de doi solvenți, în
care se introduce o cantitate mică de substanță (spectral activă) care prezintă bandă de absorbție sau
de fluorescență în domeniul în care solvenții sunt transparenți. Modelul este aplicabil lichidelor ale
căror molecule pot fi considerate sferice și izotrope și permite estimarea energiilor de interacțiune
dintre molecule.
I.2.1. Modelul statistic al soluțiilor cu trei componente
Se utilizează un amestec de doi solvenți transparenți în domeniul în care se înregistrează
spectrul, iar în amestecul celor doi solvenți se introduce o cantitate mică de substanță care prezintă
bandă de absorbție (sau de fluorescență) situată în domeniul în care solvenții sunt transparenți.
Moleculele unuia dintre solvenți, pe care îl numim solvent activ, se concentrează în jurul
moleculelor spectral active. Deplasările de frecvență din spectrele electronice sunt determinate de
interacțiunile care au loc între moleculele solventului activ și moleculele spectral active.
Distanța dintre moleculele spectral active este foarte mare, deoarece concentrația acestei
substanțe în soluție este foarte mică. Astfel, soluția ternară poate fi considerată ca fiind formată din
subsisteme, fiecare subsistem fiind format, la rândul său, dintr-o moleculă spectral activă (u) și un
amestec de molecule de solvent activ (v1) și solvent inactiv (v2), sferice și izotrope, dispuse în jurul
moleculei spectral active în straturi sferice concentrice numite straturi de solvatare. Notând cu x1 și
x2 fracțiunile moleculare ale celor doi solvenți în întreaga soluție, acestea îndeplinesc condiția:
x1 + x2 = 1 (I.11)
Se neglijează interacțiunea moleculei spectral active cu moleculele de solvent din
straturile de solvatare superioare, astfel încât soluția poate fi reprezentată prin subsisteme alcătuite
dintr-o moleculă spectral activă și moleculele de solvent din primul strat de solvatare.
Se exprimă probabilitatea de ocupare a primului strat de solvatare cu N1 molecule de tip v1
și N2 molecule de tip v2 prin relația:
( )
( )
( )
(I.12)
în care ZN este suma statistică; µ1, µ2 sunt potențialele chimice ale moleculelor de tip v1, respectiv
v2; w1, w2 reprezintă energiile de interacțiune dintre o moleculă u și o moleculă v1, respectiv v2.
Având în vedere faptul că N1 și N2 îndeplinesc condiția (I.13), potențialele chimice în cazul unor
soluții ideale sunt exprimate prin relațiile (I.14) iar suma statistică este dată de relația (I.15), se
obține pentru probabilitatea P(N1, N2) expresia (I.16).
N1 + N2 = N (I.13)
; (I.14)
(
)
(I.15)
( )
(I.16)
Mărimile p1 și p2 reprezintă numărul relativ mediu de molecule de tipul v1, respectiv v2,
din primul strat de solvatare:
;
(I.17)
p1 + p2 = 1 (I.18)
;
(I.19)
Numărul de molecule de tipul v1 și v2 diferă de la un subsistem la altul, astfel încât și
energia de interacțiune dintre molecula spectral activă și moleculele solventului diferă de la un
Page 13
13
subsistem macrocanonic la altul. Prin urmare, poziția benzii electronice de absorbție este
determinată de numărul mediu de molecule de solvent de un anumit tip aflate în primul strat de
solvatare.
Energia medie de interacțiune a moleculelor primului strat de solvatare este dată de
relația: ⟨ ⟩ (I.20)
În această relație, w1 și w2 reprezintă energiile de interacțiune dintre o moleculă u și o moleculă v1,
respectiv v2 iar W12 reprezintă energia medie totală de interacțiune dintre moleculele de solvent.
Dacă W1 = Nw1 și W2 = Nw2 reprezintă energiile de interacțiune dintre molecula u și
moleculele primului strat de solvatare, dacă acesta ar fi alcătuit numai din molecule de tipul v1 sau
numai din molecule de tipul v2, obținem: ⟨ ⟩ (I.21)
Scriem relația anterioară în starea fundamentală și în starea excitată: ⟨ ⟩ (I.22)
⟨ ⟩ (I.23)
și înlocuim p2 = 1 – p1, obținând:
⟨ ⟩ ( ) (I.24)
⟨ ⟩ ( ) (I.25)
Dacă timpii de relaxare ai lichidelor au valori mari, configurația moleculelor de solvent
din cadrul unui subsistem nu se schimbă în timpul tranziției. Pe de altă parte, utilizându-se solvenți
transparenți în domeniul spectral cercetat, moleculele solvenților rămân în stare fundamentală.
Deplasările benzii electronice de absorbție la trecerea din starea de gaz ideal (când
moleculele nu interacționează între ele) în soluție ternară (substanță spectral activă + solvent activ 1
+ solvent inactiv 2) , soluție binară 1 (substanță spectral activă + solvent activ 1) sau soluție binară 2
(substanță spectral activă + solvent inactiv 2) sunt ilustrate în Fig. I.1 și exprimate prin relațiile:
( ) ⟨ ⟩ ⟨ ⟩ (I.26)
( ) (I.27)
( ) (I.28)
Obținem expresia:
(I.29)
din care, înlocuind p1, rezultă o relație care exprimă dependența dintre variația numărului de undă
din maximul benzii electronice de absorbție și fracția moleculară x1 a solventului activ în întreaga
soluție:
(I.30)
Fracția molară a solventului (1) în soluția binară este dată de expresia:
(I.31)
în care C1, ρ1 și M1 reprezintă concentrația, densitatea și, respectiv, masa molară a solventului activ.
Se trasează graficul mărimii
în funcție de
și, din intersecția cu ordonata, se
calculează diferența w2 – w1 (fără să cunoaștem natura interacțiunilor care determină valorile w1 și
w2). Dacă moleculele u și v1 au momente dipolare permanente iar moleculele v2 nu au moment
dipolar permanent, diferența w2 – w1 poate oferi informații asupra tăriei interacțiunilor dipolare
dintre moleculele de tip u și v1.
Page 14
14
Fig. I.1. Deplasarea nivelurilor energetice la trecerea moleculei spectral active
din starea de gaz în soluții ternare și binare
I.2.2. Manifestări ale interacțiunilor de dispersie din starea lichidă
în spectrele electronice de absorbție
Există două tipuri de interacțiuni care se exercită între moleculele aflate în starea lichidă:
interacțiuni locale (specifice sau cvasichimice) și interacțiuni de tip van der Waals. Interacțiunile
locale se manifestă între două sau trei molecule ce formează sisteme moleculare relativ stabile.
Aceste interacțiuni contribuie la formarea legăturii de hidrogen sau a complecșilor moleculari cu
transfer de sarcină. Interacțiunile de tip van der Waals (universale) sunt nesaturate, se exercită între
un număr mare de molecule și pot fi, la rândul lor, interacțiuni de orientare, de inducție și de
dispersie.
Raza de acțiune a interacțiunilor universale este de câteva diametre moleculare, în sfera de
atracție moleculară. Forțele de dispersie pot fi evaluate numai în limitele mecanicii cuantice, ele
neavând echivalent în electrodinamica clasică. În lichidele cu molecule neutre și nepolare,
predominante sunt interacțiunile de dispersie. Datorită mișcării electronilor, moleculele neutre și
nepolare capătă momente dipolare instantanee și induc, în moleculele vecine, momente dipolare.
Forțele de interacțiune dintre aceste sisteme atomice neutre cu momente dipolare instantanee au fost
numite forțe de dispersie. Potențialul interacțiunii de dispersie depinde de potențialele de ionizare I1
și I2 ale moleculelor, de polarizabilitățile α1 și α2 ale acestora și de distanța r dintre centrele
moleculelor:
( )
(I.32)
Potențialul interacțiunii de dispersie poate fi scris și sub forma:
( )
(I.33)
unde ϑ01 și ϑ02 sunt frecvențele maxime din spectrul electronic de linii (sau de benzi) sau frecvențele
minime din spectrul continuu al ionului respectiv. În cazul sistemelor atomice identice, expresia
potențialului de interacțiune prin forțe dispersive devine:
Page 15
15
( )
(I.34)
Se introduce o substanță spectral activă în concentrație foarte mică într-un solvent care
trebuie să fie transparent în domeniul spectral cercetat. Moleculele trebuie să fie neutre și nepolare.
Se presupune că potențialul dispersiv este aditiv. Soluția poate fi considerată ca fiind formată din
subsisteme, fiecare subsistem fiind format dintr-o moleculă spectral activă (notată cu u), înconjurată
de moleculele de solvent (notate cu v) din sfera ei de atracție moleculară. Numărul moleculelor de
solvent dintr-un strat sferic cuprins între două sfere de raze r și r + dr este:
(I.35)
iar energia de interacțiune dintre o moleculă spectral activă și moleculele de solvent cuprinse în
acest strat este:
( )
(I.36)
Utilizând următoarea relație dintre polarizabilitatea solventului și raza moleculelor acestuia:
(I.37)
unde
( ) (I.38)
reprezintă funcția de dispersie, care depinde de indicele de refracție al solventului, și integrând după
r între limitele ru și ∞, obținem potențialul total de interacțiune prin forțe de dispersie între molecula
spectral activă și moleculele de solvent din sfera de solvatare:
( ) ∫ ( )
(I.39)
În această relație, αu și ru reprezintă polarizabilitatea și, respectiv, raza moleculei spectral active, Iu
și Iv reprezintă potențialul de ionizare al moleculei spectral active și, respectiv, al moleculei de
solvent.
Din studiul spectrelor electronice de absorbție se obțin informații despre tăria
interacțiunilor van der Waals. Dacă moleculele solvitului și solventului sunt neutre și nepolare, prin
studiul acestor spectre se pot estima interacțiunile de dispersie. În starea de vapori, interacțiunile
dintre molecule sunt neglijabile. La trecerea din starea de vapori în starea lichidă, interacțiunile
dintre molecule determină deplasarea benzilor electronice de absorbție. În cazul moleculelor sferice
neutre și nepolare, deplasarea nivelurilor energetice este datorată, în primul rând, interacțiunilor de
dispersie.
În Fig. I.2 este ilustrată deplasarea nivelurilor energetice la trecerea din starea de gaz ideal
în stare lichidă. Indicii g și e se referă la stările fundamentală și excitată ale moleculei spectral
active. Variația numărului de undă la trecerea moleculei din starea de vapori în starea lichidă, din
cauza interacțiunilor de dispersie, este dată de relația:
( )
(
)
(I.40)
Interacțiunile dintre moleculele spectral active au fost neglijate deoarece spectrele electronice se
înregistrează la concentrații foarte mici ale substanței spectral active (10-3 – 10-5 mol/l) iar distanțele
dintre aceste molecule sunt mari. S-a presupus că, prin excitare, polarizabilitatea moleculei spectral
active se poate modifica. Relația (I.40) arată că variația numărului de undă din maximul benzii
electronice de absorbție, la trecerea din starea de gaz în cea de soluție, este proporțională cu funcția
de dispersie a solventului. Aceeași relație arată că temperatura nu influențează tăria interacțiunilor
de dispersie. Influența interacțiunilor de dispersie asupra deplasărilor spectrale poate fi demonstrată
prin înregistrarea spectrelor electronice de absorbție ale unei molecule spectral active nepolare
solvită într-un solvent nepolar.
Page 16
16
Fig. I.2. Deplasarea nivelurilor energetice la trecerea moleculei spectral active
din starea de gaz ideal în starea lichidă, ca urmare a interacțiunilor de dispersie
Page 17
17
Partea a II-a. Contribuții personale
Capitolul II. Caracterizarea cuanto-mecanică și solvatocromică a quercetinei
II.1. Introducere
Flavonoidele sunt molecule care se găsesc în multe fructe și legume, fiind cei mai
importanți pigmenți din plante, responsabili pentru colorarea florilor. Sunt compuși polifenolici cu
structura generală a unui schelet format din 15 atomi de carbon, având două inele fenil și un inel
heterociclic (C6 – C3 – C6) (vezi Fig. II.1). În plantele care se găsesc la înălțimi mai mari,
flavonoidele sunt implicate în filtrarea ultravioletelor, fixarea azotului simbiotic și pigmentarea
florală [1].
Quercetina este un flavonoid de tipul flavonolilor (cei mai abundenți dintre moleculele de
flavonoizi), găsit într-o varietate de alimente incluzând fructe, legume, semințe și nuci. Quercetina
este, de asemenea, prezentă în vinul roșu și în diferite tipuri de miere [2].
Quercetina este un candidat promițător în terapia cancerului deoarece inhibă proliferarea și
proprietățile canceroase ale adenocarcinomului de colon și ale celulelor adenom precoce [12].
Quercetina conjugată cu nanoparticule de nichel acoperite cu polietilen glicol poate fi folosită ca un
agent chimioterapeutic promițător împotriva celulelor cancerului de sân. Studiile suplimentare pot
evalua activitatea anticanceroasă a quercetinei pe diferite tipuri de celule canceroase [17].
Chemoprevenirea prin modificarea regimului alimentar (creșterea consumului de alimente pe bază
de plante) pare a fi cea mai promițătoare și rentabilă metodă de a reduce riscul de cancer.
Quercetina pare să aibă efecte antivirale directe și mediate de gazdă, împotriva
următoarelor virusuri: virusul hepatitei C [19], virusul gripal AH1N1 [20], virusul Herpex Simplex
tip 1, poliovirus tip 1 și virus sincițial respirator (RSV/VSR). De asemenea, quercetina are activitate
antibacteriană in vitro împotriva bacteriei Helicobacter pylori și a cinci microorganisme asociate cu
debutul și progresia bolii parodontale [5].
Quercetina poate avea aplicații clinice în gestionarea și prevenirea complicațiilor
neurologice (precum disfuncția memoriei mediată de stres) asociate cu stresul cronic și diabetul de
tip 2 [25]. Tratamentul cu quercetină a atenuat rezistența la insulină. Quercetina poate oferi o
abordare promițătoare pentru tratamentul bolii Alzheimer și a altor boli neurodegenerative asociate
stresului oxidativ [26, 27] și ar putea ameliora depresia asociată cu epilepsie, precum și depresia
comorbidă [28].
Quercetina acționează ca un potențial agent protector împotriva acrilamidei (o substanță
asociată cu riscurile de cancer, formată în alimentele bogate în carbohidrați, în timpul preparării la
temperaturi înalte) [30] și furanului (un agent cancerigen din ficat, produs în timpul procesului de
încălzire a alimentelor) [31].
Quercetina posedă proprietăți anti-îmbătrânire și de întinerire, având un potențial mare de
a fi utilizată în produsele naturale anti-îmbătrânire [33].
Flavonoidele conțin mai multe grupări hidroxil cu activitate de captare a radicalilor liberi
printr-un proces de donare de electroni. Structura cu două grupări hidroxil adiacente are o capacitate
sporită de donare de electroni. Quercetina, ca și alte flavonoide care conțin structura catecolică,
prezintă activitate antiradicală importantă [35].
Anumiți parametri fizico-chimici ai moleculelor spectral active pot fi evaluați prin studii
solvatocromice atunci când procesul de solvatare induce schimbări în poziția benzilor electronice de
absorbție. Au fost stabilite corelații teoretice [36-38] și empirice [39] între numărul de undă din
maximul benzii electronice de absorbție a moleculei spectral active și parametrii solventului
(indicele de refracție și permitivitatea electrică) pentru a descrie influența interacțiunilor universale
Page 18
18
(inducție, orientare, polarizare și dispersie) asupra poziției benzii electronice. Interacțiunile specifice
dintre moleculele de solvit și cele de solvent (neglijate în teoria soluțiilor) pot fi evidențiate utilizând
parametrii microscopici ai solventului, cum ar fi α și β, care sunt proporționali cu tăria legăturilor de
hidrogen, realizate de solvent ca donor sau, respectiv, acceptor de protoni.
Prin combinarea rezultatelor teoretice obținute de McRae [36] și modificate de alți
cercetători [38, 40-42] pentru deplasările spectrale ale benzilor electronice de absorbție datorate
interacțiunilor universale, cu expresiile empirice pentru interacțiunile specifice introduse de Kamlet
și Taft, se poate obține o relație de tipul:
( ) ( ) (II.1)
unde este numărul de undă corespunzător maximului benzii de absorbție analizate, are
semnificația numărului de undă din maximul benzii electronice de absorbție pentru faza gazoasă a
substanței spectral active, C1 – C4 sunt coeficienții de regresie obținuți prin analiză de regresie
liniară multiplă, f(ε) și f(n) reprezintă funcția de polaritate a solventului și, respectiv, funcția de
polarizabilitate a solventului (funcția de dispersie), fiind date de relațiile:
( )
(II.2)
( )
(II.3)
iar α și β sunt parametrii Kamlet-Taft ai solventului (α –donor în legătura de hidrogen, β – acceptor
în legătura de hidrogen).
Relația (II.1) poate fi supusă foarte ușor unei analize statistice când banda electronică a
moleculei spectral active este înregistrată într-un număr mare de solvenți având proprietăți fizico-
chimice diferite. Coeficienții de regresie C1 – C4 dau informații despre mărimea deplasării spectrale
cauzate de fiecare tip de interacțiune și despre sensul deplasării în scala numerelor de undă a
radiațiilor electromagnetice. Analiza statistică poate fi efectuată în condițiile în care valorile
mărimilor (cm-1), ε , n, β și α sunt cunoscute pentru toți solvenții utilizați. Termenii C1f(ε) și C2f(n)
exprimă contribuțiile interacțiunilor universale prin procese de orientare-inducție și, respectiv,
polarizare-dispersie. Termenii C3β și C4α exprimă contribuția interacțiunilor specifice în care
molecula spectral activă (solut) donează și, respectiv, acceptă protoni.
Informațiile referitoare la efectele solvatocromice au fost aplicate quercetinei, o moleculă
spectral activă cu o bandă spectrală de natură π – π* bine individualizată, care se deplasează spre
roșu când polaritatea solventului crește. Au fost obținute, prin studiu solvatocromic, informații
despre polarizabilitatea și momentul de dipol ale moleculei de quercetină în stare excitată.
II.2. Detalii computaționale și experimentale
Quercetina (sau 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one) a fost
procurată de la Sigma-Aldrich Chemical Company (având o puritate (HPLC) ≥ 95%) și a fost
utilizată fără purificare suplimentară. Solvenții, de calitate spectrală (cei mai mulți dintre ei, ACS
reagent sau Reagent Plus, cu puritate > 99%), au fost achiziționați de la Merck. Quercetina este o
pulbere cristalină de culoare galbenă, practic insolubilă în apă, solubilă în soluții apoase alcaline.
Densitatea ei este de 1.799 g/cm3 iar punctul de topire este de 316 C.
Spectrele electronice de absorbție ale quercetinei au fost înregistrate în 15 solvenți la
temperatura camerei, cu un spectrometru Ocean-Optics QE 65000 UV-Vis. Analiza statistică a
datelor solvatocromice a fost realizată utilizând programul Origin 8.
Anumite proprietăți moleculare ale quercetinei au fost calculate utilizând programul
Spartan’14 [43]. Programul Spartan este un pachet de modelare moleculară utilizat în chimie.
Interfața Spartan oferă modele computaționale moderne, inclusiv modele de mecanică moleculară,
Page 19
19
modele orbitale moleculare semi-empirice și Hartree-Fock, precum și o varietate de modele corelate,
inclusiv modele funcționale de densitate și modele Møller-Plesset.
Modelele semi-empirice sunt cele mai simple modele bazate pe mecanica cuantică. Sunt
aplicabile moleculelor conținând 100-200 de atomi. Aceste modele oferă, în general, geometrii în
bună concordanță cu structura experimentală. Modelele semi-empirice sunt, în general, adecvate
pentru evaluarea unor proprietăți (precum aria suprafeței polare) care depind doar de geometrie.
Modelele semi-empirice sunt disponibile pentru calcularea spectrelor din infraroșu, dar nu
furnizează un rezultat foarte bun. Nu sunt disponibile pentru calculul spectrelor Raman, spectrelor
de rezonanță magnetică nucleară sau spectrelor din ultraviolet și vizibil.
Modelele Hartree-Fock rezultă din ecuația Schrödinger prin impunerea cerinței ca
electronii să fie particule independente. Modelele Hartree-Fock sunt disponibile pentru calculul
spectrelor din infraroșu, spectrelor Raman, spectrelor de rezonanță magnetică nucleară și a celor din
ultraviolet sau vizibil. Modelele care utilizează aproximația Hartree-Fock sunt denumite modele
corelate. Acestea se împart în două mari categorii: modele funcționale de densitate și modele bazate
pe funcția de undă. Modelele funcționale de densitate și modelele Møller-Plesset asigură un calcul
excelent al geometriilor de echilibru ale moleculelor organice și sunt, în general, de preferat
modelelor Hartree-Fock în acest scop. Modelele funcționale de densitate sunt disponibile pentru
calculul spectrelor din infraroșu, Raman, RMN și ultraviolet/vizibil, ultimele în conjuncție cu așa-
numitele modele funcționale de densitate dependente de timp pentru calculele care implică stări
excitate.
II.3. Rezultate și discuții
II.3.1. Caracterizare cuanto-mecanică a quercetinei
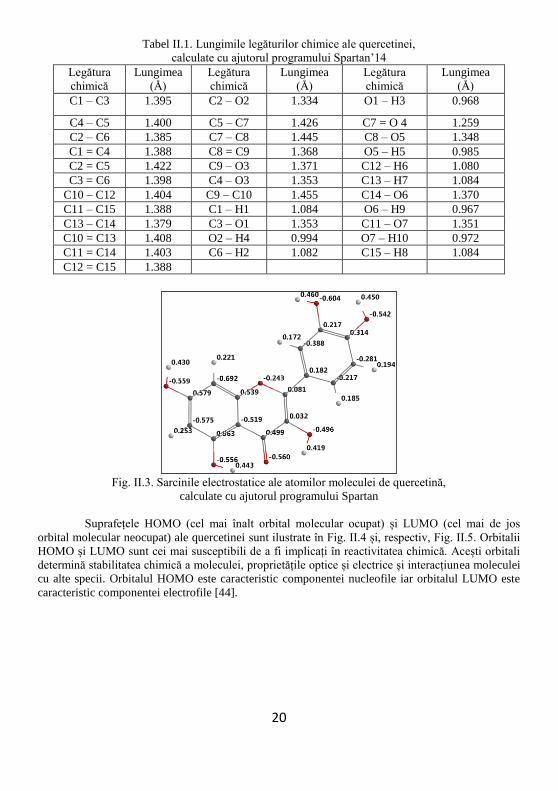
Structura moleculară a quercetinei este ilustrată în Fig. II.1, unde este reprezentat și
vectorul moment de dipol al moleculei. Structura moleculară a fost optimizată cu ajutorul
programului Spartan’14, utilizând metoda funcționalei de densitate.
Programul Spartan furnizează distanțele dintre atomii moleculei, unghiurile dintre legături,
precum și unghiurile diedre. Lungimile legăturilor covalente ale moleculei de quercetină sunt listate
în Tabelul II.1. Atomii moleculei sunt etichetați în Fig. II.2.
Sarcinile electrostatice ale atomilor moleculei, calculate cu ajutorul programului de
modelare moleculară Spartan’14 și exprimate în procente ale sarcinii electrice elementare, sunt
afișate în Fig. II.3.
Fig. II.1. Structura chimică a
quercetinei (C – gri, O – roșu, H – alb)
Fig. II.2. Structura chimică a
quercetinei, cu atomii etichetați
Page 20
20
Tabel II.1. Lungimile legăturilor chimice ale quercetinei,
calculate cu ajutorul programului Spartan’14
Legătura
chimică
Lungimea
(Å)
Legătura
chimică
Lungimea
(Å)
Legătura
chimică
Lungimea
(Å)
C1 – C3 1.395 C2 – O2 1.334 O1 – H3 0.968
C4 – C5 1.400 C5 – C7 1.426 C7 = O 4 1.259
C2 – C6 1.385 C7 – C8 1.445 C8 – O5 1.348
C1 = C4 1.388 C8 = C9 1.368 O5 – H5 0.985
C2 = C5 1.422 C9 – O3 1.371 C12 – H6 1.080
C3 = C6 1.398 C4 – O3 1.353 C13 – H7 1.084
C10 – C12 1.404 C9 – C10 1.455 C14 – O6 1.370
C11 – C15 1.388 C1 – H1 1.084 O6 – H9 0.967
C13 – C14 1.379 C3 – O1 1.353 C11 – O7 1.351
C10 = C13 1.408 O2 – H4 0.994 O7 – H10 0.972
C11 = C14 1.403 C6 – H2 1.082 C15 – H8 1.084
C12 = C15 1.388
Fig. II.3. Sarcinile electrostatice ale atomilor moleculei de quercetină,
calculate cu ajutorul programului Spartan
Suprafețele HOMO (cel mai înalt orbital molecular ocupat) și LUMO (cel mai de jos
orbital molecular neocupat) ale quercetinei sunt ilustrate în Fig. II.4 și, respectiv, Fig. II.5. Orbitalii
HOMO și LUMO sunt cei mai susceptibili de a fi implicați în reactivitatea chimică. Acești orbitali
determină stabilitatea chimică a moleculei, proprietățile optice și electrice și interacțiunea moleculei
cu alte specii. Orbitalul HOMO este caracteristic componentei nucleofile iar orbitalul LUMO este
caracteristic componentei electrofile [44].
Page 21
21
Fig. II.4. Suprafața HOMO Fig. II.5. Suprafața LUMO
Anumite proprietăți moleculare ale quercetinei (energia, energia în apă, energia de
solvatare, momentul de dipol, aria, volumul, polarizabilitatea), determinate cu ajutorul programului
Spartan’14, sunt prezentate în Tabelul II.2.
Tabel II.2. Proprietăți moleculare și energii calculate ale quercetinei
Formula chimică C15H10O7
Masa moleculară 302,238 amu
Energia -1103.51 au
Energia în apă -1103.54 au
Energia de solvatare -67.35 kJ/mol
EHOMO -5.36 eV
ELUMO -1.92 eV
Momentul de dipol 5.70 Debye
Tautomeri 3
Conformeri 64
Relația cantitativă structură – activitate
Aria 276.05 Å2
Volumul 264.19 Å3
Aria suprafeței polare 105.755 Å2
Ovalitate 1.39
Log P
P – coeficient de partiție
-4.54
Polarizabilitate 61.99
Numărul donorilor în legătura de hidrogen 5
Numărul acceptorilor în legătura de hidrogen 7
Temperatura 298.15 K
ΔE = |EHOMO - ELUMO| 3.44 eV
Potențialul de ionizare , I 5.36 eV
Afinitatea electronică , A 1.92 eV
Electronegativitatea, χ 3.64 eV
Tăria chimică, η 1.72 eV
Indicele de electrofilicitate, ω 3.85 eV
Potențialul chimic, μ -3.64 eV
Page 22
22
Diferența HOMO-LUMO (ΔE) este cea mai mică energie necesară pentru excitarea
moleculei, determinând stabilitatea chimică și reactivitatea moleculei.
ΔE = |EHOMO – ELUMO| (II.4)
Potențialul de ionizare (I) și afinitatea electronică (A) pot fi estimate utilizând teorema lui
Koopmans:
I = - E HOMO (II.5)
A = - ELUMO (II.6)
Valorile electronegativității (χ), tăriei chimice (η), indicelui de electrofilicitate (ω) și
potențialului chimic (µ) pot fi calculate cu următoarele ecuații:
(II.7)
(II.8)
(II.9)
(II.10)
Potențialul chimic (μ) este de obicei negativ și măsoară tendința de donare a electronilor.
Tăria chimică (η) măsoară rezistența la transferurile de electroni; este, de obicei, pozitivă.
O valoare absolută mare a energiei moleculare determină o stabilitate mare a moleculei.
Energia de solvatare reprezintă cantitatea de energie necesară pentru dizolvarea unui solut într-un
solvent. Procesul de dizolvare poate fi endoterm sau exoterm. Energia în apă caracterizează procesul
de soluție (dizolvare), ea reprezentând energia absorbită atunci când substanța este dizolvată în apă.
Momentul de dipol electric este o mărime fizică vectorială ce măsoară separarea sarcinilor
pozitive și negative ale unui sistem de sarcini electrice. Multe molecule au moment electric dipolar
datorită repartizării neuniforme a sarcinilor electrice pozitive și negative pe atomii constituenți.
Coeficientul de partiție P este definit ca un anumit raport al concentrațiilor unui solut în
doi solvenți (o bifază de faze lichide), în special pentru soluți neionizați [45]. Când unul dintre
solvenți este apa iar celălalt este un solvent nepolar, valoarea log P este o măsură a lipofilicității sau
hidrofobicității. În funcție de această valoare, structura chimică are caracter hidrofil atunci când log
P < 0 și hidrofob atunci când log P > 0 [46].
Aria suprafeței polare a unei molecule se definește ca suma suprafețelor atomilor polari
(de obicei oxigen, azot și hidrogen atașat) dintr-o moleculă. Acest parametru este foarte util pentru
predicția proprietăților transportului de medicamente. Membranele celulare pot fi traversate doar de
moleculele cu aria suprafeței polare mai mică decât 140 Å2. De asemenea, bariera hemato-encefalică
poate fi penetrată numai de către moleculele pentru care aria suprafeței polare este mai mică decât
90 Å2 [47]. Aria suprafeței polare a quercetinei este 105,75 Å2. Prin urmare, aceste molecule pot
traversa membranele celulare, dar nu pot penetra bariera hemato-encefalică.
Programul Spartan furnizează importante reprezentări grafice care rezultă din calcule de
chimie cuantică. Harta potențialului electrostatic descrie distribuția globală a sarcinii moleculare și
ajută la anticiparea regiunilor de adiție electrofilă (Fig. II.6). Culorile spre roșu corespund
potențialului negativ, în timp ce culorile spre albastru corespund potențialului pozitiv. Harta
potențialului de ionizare locală este un alt indicator al adiției electrofile (Fig. II.7). Culorile spre roșu
corespund potențialelor de ionizare mici iar culorile spre albastru corespund potențialelor de
ionizare mari. Harta |LUMO| este un indicator al adiției nucleofile (Fig. II.8). Prin convenție,
culorile spre roșu indică valori absolute mici ale LUMO (apropiate de zero), în timp ce culorile spre
albastru indică valori absolute mari ale LUMO [43].
După cum rezultă din Fig. II.6, potențialul electrostatic are valori intermediare pe aproape
întreaga suprafață a moleculei de quercetină, excepție făcând zonele periferice, unde potențialul
electrostatic are valoarea cea mai mică, negativă (în jurul atomilor de oxigen), respectiv valoarea cea
mai mare, pe atomii de hidrogen. Harta potențialului de ionizare locală (Fig. II.7) indică doar valori
Page 23
23
ridicate și intermediare ale acestui potențial. De asemenea, harta |LUMO| (Fig. II.8) ilustrează valori
mici, apropiate de zero ale LUMO, pe cea mai mare parte a suprafeței moleculei de quercetină.
Fig. II.6. Harta potențialului Fig. II.7. Harta potențialului Fig. II.8. Harta |LUMO|
electrostatic de ionizare locală
II.3.2. Rezultate spectrale
Spectrele electronice de absorbție ale quercetinei au fost înregistrate în 15 solvenți, la
temperatura camerei. În Fig. II.9 sunt prezentate câteva dintre benzile electronice de absorbție din
ultraviolet și vizibil ale quercetinei.
200 300 400 500 600
0.0
0.2
0.4
0.6
0.8
1.0
Ab
so
rban
ta (
u.a
.)
Lungimea de unda (nm)
Quercetina + Metanol
200 300 400 500 600
0.0
0.1
0.2
0.3
0.4
0.5
0.6
Absorb
anta
(u.a
.)
Lungimea de unda (nm)
Quercetina + Acetonitril
200 300 400 500 600
0.0
0.1
0.2
0.3
0.4
0.5
Ab
so
rban
ta (
u.a
.)
Lungimea de unda (nm)
Quercetina + Etanol
Fig. II.9. Benzile electronicele de absorbție ale quercetinei, în ultraviolet și vizibil
A fost utilizată dependența multiliniară exprimată prin relația (II.1) pentru a descrie
deplasările spectrale ale quercetinei, măsurate în solvenții utilizați. Datele spectroscopice au fost
procesate utilizând analiza de regresie liniară multiplă. Valorile coeficienților ce intervin în relația
(II.1) sunt listate în Tabelul II.5.
Tabel II.5. Coeficienții de regresie liniară
( ) C1
( ) C2 (
) C3 ( ) C4 (
) Coeficientul
de corelație R
Deviația
standard SD
29906.63 -652.40 -7457.04 -510.09 -221.37 0.96 54.22
Valorile experimentale ale numărului de undă din maximul benzii π → π* și, de asemenea,
valorile calculate ale numărului de undă pe baza relației (II.1), cu coeficienții de corelație obținuți
prin analiză statistică, se regăsesc în Tabelul II.6. Relația obținută este de tipul:
( ) ( ) (II.11)
Page 24
24
Tabel II.6. Numerele de undă experimentale ( ) și calculate ( ) pentru solvenții utilizați;
funcția de polaritate a solventului, f(ε); funcția de polarizabilitate a solventului, f(n); β – capacitatea
de a accepta protoni; α – capacitatea de a dona protoni.
Nr. Solvent
( )
( ) f (n)
( )
1 Metanol 27291.83 0.914 0.203 0.66 0.98 27242.96
2 Acetonitril 27525.46 0.924 0.212 0.40 0.19 27476.82
3 Etanol 27062.13 0.887 0.221 0.75 0.86 27107.00
4 Acetat de etil 27525.46 0.626 0.227 0.45 0.00 27575.94
5 2-Propanol 27118.64 0.863 0.230 0.84 0.76 27031.77
6 2-Butanonă 27350.05 0.854 0.231 0.48 0.06 27368.78
7 1-Propanol 26948.37 0.866 0.235 0.90 0.84 26944.22
8 2-Butanol 26948.37 0.838 0.240 0.80 0.69 27009.41
9 1-Butanol 26948.37 0.846 0.242 0.84 0.84 26935.67
10 1-Hexanol 26891.84 0.804 0.252 0.84 0.80 26897.35
11 Etilenglicol 26836.27 0.924 0.258 0.52 0.90 26915.42
12 Formamidă 26891.84 0.965 0.267 0.48 0.71 26884.02
13 Dimetilsulfoxid 26780.21 0.938 0.284 0.76 0.00 26789.21
14 Anisol 27176.13 0.526 0.303 0.32 0.00 27140.76
15 Alcool benzilic 26669.51 0.800 0.314 0.52 0.60 26645.13
Coeficienții de regresie din relația (II.11) sunt negativi, indicând deplasări spectrale spre
roșu atunci când parametrii ε, n, β și α ai solventului cresc. Acest rezultat este în concordanță cu
datele experimentale din Tabelul II.6.
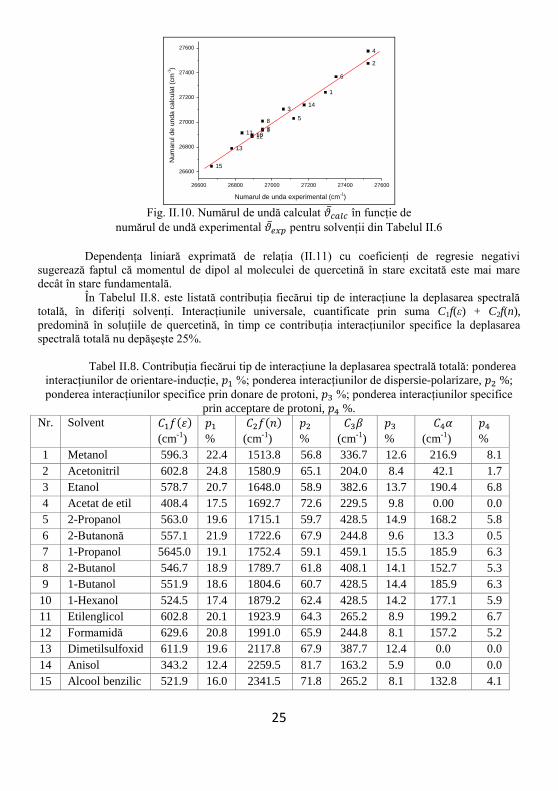
Dependența numărului de undă calculat, (cm-1), în funcție de numărul de undă
experimental, (cm-1), este reprezentată grafic în Fig. II.10. Există o dependență liniară
caracterizată de panta 0.97 (eroarea fiind de 0.05) iar coeficientul de regresie este R = 0.97 (vezi
Tabelul II.7). Deviația standard este SD = 46.82. Există o bună corelație liniară între valorile
calculate și cele experimentale ale numerelor de undă din maximele benzilor din vizibil ale
quercetinei.
Tabel II.7. Caracteristicile dependenței liniare dintre
numărul de undă calculat și numărul de undă experimental
Intersecția cu ordonata Panta dreptei Date statistice
Valoare Eroare Valoare Eroare Coeficientul de
corelație R
Deviația standard
SD
826.53 1291.70 0.97 0.05 0.97 46.82
Page 25
25
26600 26800 27000 27200 27400 27600
26600
26800
27000
27200
27400
27600
1
2
3
4
5
6
7
8
9101112
13
14
15N
um
aru
l d
e u
nd
a c
alc
ula
t (c
m-1)
Numarul de unda experimental (cm-1)
Fig. II.10. Numărul de undă calculat în funcție de
numărul de undă experimental pentru solvenții din Tabelul II.6
Dependența liniară exprimată de relația (II.11) cu coeficienți de regresie negativi
sugerează faptul că momentul de dipol al moleculei de quercetină în stare excitată este mai mare
decât în stare fundamentală.
În Tabelul II.8. este listată contribuția fiecărui tip de interacțiune la deplasarea spectrală
totală, în diferiți solvenți. Interacțiunile universale, cuantificate prin suma C1f(ε) + C2f(n),
predomină în soluțiile de quercetină, în timp ce contribuția interacțiunilor specifice la deplasarea
spectrală totală nu depășește 25%.
Tabel II.8. Contribuția fiecărui tip de interacțiune la deplasarea spectrală totală: ponderea
interacțiunilor de orientare-inducție, %; ponderea interacțiunilor de dispersie-polarizare, %;
ponderea interacțiunilor specifice prin donare de protoni, %; ponderea interacțiunilor specifice
prin acceptare de protoni, %.
Nr. Solvent ( )
(cm-1)
%
( )
(cm-1)
%
(cm-1)
%
(cm-1)
%
1 Metanol 596.3 22.4 1513.8 56.8 336.7 12.6 216.9 8.1
2 Acetonitril 602.8 24.8 1580.9 65.1 204.0 8.4 42.1 1.7
3 Etanol 578.7 20.7 1648.0 58.9 382.6 13.7 190.4 6.8
4 Acetat de etil 408.4 17.5 1692.7 72.6 229.5 9.8 0.00 0.0
5 2-Propanol 563.0 19.6 1715.1 59.7 428.5 14.9 168.2 5.8
6 2-Butanonă 557.1 21.9 1722.6 67.9 244.8 9.6 13.3 0.5
7 1-Propanol 5645.0 19.1 1752.4 59.1 459.1 15.5 185.9 6.3
8 2-Butanol 546.7 18.9 1789.7 61.8 408.1 14.1 152.7 5.3
9 1-Butanol 551.9 18.6 1804.6 60.7 428.5 14.4 185.9 6.3
10 1-Hexanol 524.5 17.4 1879.2 62.4 428.5 14.2 177.1 5.9
11 Etilenglicol 602.8 20.1 1923.9 64.3 265.2 8.9 199.2 6.7
12 Formamidă 629.6 20.8 1991.0 65.9 244.8 8.1 157.2 5.2
13 Dimetilsulfoxid 611.9 19.6 2117.8 67.9 387.7 12.4 0.0 0.0
14 Anisol 343.2 12.4 2259.5 81.7 163.2 5.9 0.0 0.0
15 Alcool benzilic 521.9 16.0 2341.5 71.8 265.2 8.1 132.8 4.1
Page 26
26
Coeficienții de regresie C1 și C2 din ecuația (II.1) depind de parametrii microscopici ai
moleculei de solut, astfel [48,49]:
( )
(II.12)
( )
(II.13)
Ecuațiile (II.12) și (II.13) permit estimarea momentului de dipol și a polarizabilității
moleculei spectral active, în stare excitată. Însumând relațiile (II.12) și (II.13), obținem:
(II.14)
Raza moleculară a poate fi calculată utilizând valorile volumului și ariei suprafeței, în
starea fundamentală a moleculei respective [42]:
(II.15)
În ecuațiile (II.12)-(II.14) au fost făcute următoarele notații: µg și µe sunt momentele de
dipol ale moleculei de quercetină în starea fundamentală și, respectiv, în starea excitată, φ este
unghiul dintre aceste momente de dipol, αg și αe sunt polarizabilitățile moleculei spectral active în
starea fundamentală și, respectiv, în starea excitată, c este viteza luminii în vid, a este raza
moleculei, h reprezintă constanta lui Planck, k - constanta lui Boltzmann, T - temperatura absolută,
Iu - potențialul de ionizare al moleculei spectral active, Iv - potențialul de ionizare al moleculei de
solvent. Solventul ales pentru calculul polarizabilității și momentului de dipol în starea excitată a
moleculelor spectral active a fost dimetilsulfoxid.
Polarizabilitatea în starea excitată a moleculei spectral active poate fi exprimată prin
ecuația (II.16), așa cum rezultă din relația (II.14):
(II.16)
Utilizând relațiile (II.16) și (II.12), se obține:
(II.17)
Pentru ca ecuația (II.17) să aibă soluții reale, discriminantul ei trebuie să fie pozitiv:
(II.18)
Rezultă că unghiul φ trebuie să satisfacă inegalitatea: φ < 78.81 . În acest caz, valorile
acceptate ale momentului de dipol în stare excitată sunt cuprinse între următoarele limite:
(II.19)
Limitarea suplimentară a valorii momentului de dipol al moleculei de quercetină în stare
excitată, bazată pe studiul solvatocromic, poate fi făcută considerând că deplasările spectrale indică
valabilitatea relațiilor (II.16) și (II.17) pentru polarizabilitatea moleculei spectral active și momentul
de dipol în stare excitată. În concordanță cu aceste relații, atunci când unghiul φ (dintre momentele
de dipol ale quercetinei în stările electronice responsabile de apariția benzii studiate) crește, crește și
momentul de dipol în stare excitată, în timp ce polarizabilitatea moleculei în stare excitată descrește.
De obicei, polarizabilitatea în stare excitată este mai mare decât în stare fundamentală.
Astfel, αe pentru quercetină poate descrește până la valoarea αg = 61.99 Å3, așa cum rezultă din
calculele de mecanică cuantică. Pentru această valoare, din relația (II.16) se obține µe = 8.38 D iar
unghiul corespunzător φ este 44.58 de grade.
Deplasarea batocromică a benzii electronice de absorbție din vizibil a quercetinei, care are
loc când molecula trece din starea gazoasă în soluții cu polaritate crescătoare, rezultă din semnele
negative ale coeficienților de regresie din ecuația (II.17).
Page 27
27
II.4. Concluzii
Din studiul cuanto-mecanic rezultă că molecula de quercetină este una hidrofilă, care
poate traversa membranele celulare, dar nu poate penetra bariera hemato-encefalică.
Valoarea negativă a energiei de solvatare indică faptul că procesul de solvatare a
quercetinei este unul exoterm (cu degajare de căldură).
Studiul solvatocromic a subliniat prevalența interacțiunilor de dispersie-polarizare în
soluțiile binare ale quercetinei. Utilizând valorile coeficienților de regresie obținuți în studiul
solvatocromic, se poate afirma că momentul de dipol al quercetinei crește prin excitare de la
valoarea calculată (µg = 5.79 D) pentru molecula izolată, la µe = 8.38 D în soluția de dimetilsulfoxid.
Unghiul dintre momentele de dipol în stările moleculare participante la tranziția electronică este mai
mic decât 45 de grade. Acest studiu contribuie la o mai bună reprezentare a transformării suferite de
moleculă atunci când interacționează cu radiația electromagnetică din domeniul vizibil.
Capitolul III. Studiul computațional și solvatocromic
al moleculelor de piridiniu-acetil-benzoil-metilid
III.1. Introducere
În ilide [50,51], carbanionul, încărcat negativ, este atașat direct unui heteroatom încărcat
cu o sarcină pozitivă. Ilidele în care carbanionul este legat covalent de un atom de azot aparținând
unui heterociclu (piridiniu, izo-chinolină, piridazină, benzo[f]chinolină) sunt numite cicloimoniu-
ilide [50]. Acestea sunt substanțe puternic nucleofile care pot reacționa cu o mare varietate de
compuși organici. Datorită naturii lor dipolare, cicloimoniu-ilidele pot participa la reacții de
cicloadiție, cu reactivi dienofili, constituind o sursă de noi compuși heterociclici.
Cicloimoniu-ilidele în care cationul aparține ciclului piridinic sunt numite piridiniu-ilide.
În funcție de structura componentei anionice, piridiniu-ilidele pot fi clasificate în piridiniu-metilide,
piridiniu-metilide-carbanion-monosubstituite și piridiniu-metilide-carbanion-disubstituite (Fig.III.1).
N+
CH2
-
N+
C-
R
H
N+
C-
R1
R2
Fig. III.1. Piridiniu-metilidă (a), piridiniu-metilidă-carbanion-monosubstituită (b),
piridiniu-metilidă-carbanion-disubstituită (c)
Piridiniu-ilidele carbanion disubstituite cu grupări acceptoare de electroni sunt stabile,
fiind studiate din multe puncte de vedere. O stabilitate redusă o au piridiniu-metilidele care au o
singură grupare cu caracter electronegativ legată covalent de carbonul ilidic. Piridiniu-ilidele
carbanion disubstituite, obținute prin “metoda sării” sau prin alte căi de sinteză, sunt în general
produși cristalizați, conservabili, stabili față de oxigen și umiditate.
a) b) c)
Page 28
28
Spectrele de absorbție în ultraviolet și vizibil ale unor piridiniu-ilide contribuie la o mai
bună cunoaștere a structurii moleculare. Toate cicloimoniu-ilidele prezintă o bandă slabă în vizibil
atribuită unui transfer intramolecular de sarcină (TIS) de la carbanion către heterociclu [50,52,53].
Această bandă poate oferi informații despre natura interacțiunilor intermoleculare în soluțiile de
ilidă și, de asemenea, despre tăria acestor interacțiuni în fiecare tip de solvent. În acest scop,
cicloimoniu-ilidele sunt utilizate ca sonde dipolare în estimarea câmpului reactiv în soluții lichide
[53-56].
Mecanismul transferului intramolecular de sarcină (TIS) diminuează momentul de dipol al
solutului (chiar și sensul său poate fi schimbat în procesul TIS). Deci, interacțiunile de orientare în
starea electronică fundamentală a cicloimoniu-ilidelor sunt mai puternice decât în stările lor excitate.
Interacțiunile specifice (precum legăturile de hidrogen) [56] pot stabiliza suplimentar molecula de
ilidă în starea electronică fundamentală. În consecință, deplasările spre albastru ale benzii
electronice din vizibil au fost evidențiate pentru diferite ilide, studiate anterior, atunci când au trecut
de la solvenți nepolari la solvenți polari sau de la solvenți neprotici la solvenți protici [53-55].
Au fost determinați câțiva parametri energetici ai moleculei de piridiniu-acetil-benzoil-
metilid (PABM) și a fost analizată influența acestora asupra deplasărilor spectrale induse de solvenți
de natură diferită asupra benzii electronice de absorbție din vizibil, ca urmare a transferului
intramolecular de sarcină (TIS).
Cicloimoniu-ilidele pot participa atât la interacțiuni nespecifice cât și la interacțiuni
specifice. S-a încercat stabilirea contribuției fiecărui tip de interacțiune la deplasările spectrale totale
ale benzii de transfer intramolecular de sarcină, în diferite soluții ale PABM.
Piridiniu-acetil-benzoil-metilid este studiat aici din punct de vedere structural și
solvatocromic, pentru a stabili natura interacțiunilor intermoleculare în soluțiile sale și pentru a
estima contribuția fiecărui tip de interacțiune la deplasarea spectrală totală a benzii de transfer
intramolecular de sarcină din vizibil, înregistrată într-un solvent dat.
Rezultatul original al acestui studiu este estimarea momentului de dipol electric și
polarizabilității moleculei PABM în starea excitată, utilizând doar numerele de undă ale benzii de
transfer intramolecular de sarcină din vizibil, în diferiți solvenți, pe baza analizei statistice a
dependenței multiliniare a numărului de undă din maximul benzii de transfer intramolecular de
sarcină, de parametrii macroscopici ai solventului (n – indicele de refracție, ε – permitivitatea
electrică) și, de asemenea, de parametrii empirici β (capacitatea solventului de a accepta protoni în
legătura de hidrogen) și α (capacitatea solventului de a dona protoni în legătura de hidrogen).
Diferența dintre energiile potențiale în perechile moleculare de tipul ilidă – solvent protic
și ilidă – solvent neprotic a fost estimată în limitele modelului celular statistic al soluțiilor ternare
ale PABM. Această diferență este dificil sau chiar imposibil de măsurat prin alte metode.
Cunoașterea tăriei fiecărui tip de interacțiune în diferiți solvenți oferă informații chimiștilor despre
eficiența moleculei PABM în diferite tipuri de reacții.
III.2. Detalii computaționale și experimentale
Structura moleculară de echilibru a PABM (vezi Fig. III.3) a fost stabilită utilizând
programul Spartan’14. Câteva proprietăți moleculare ale ilidei studiate au fost calculate cu ajutorul
programului Spartan, utilizând modelul funcționalei de densitate EDF2/6-31G* (energia moleculară,
energia în apă, energia de solvatare, momentul de dipol, orbitalii de frontieră ș.a.m.d.) [43,64,65].
Page 29
29
Fig. III.3. Structura moleculară a PABM Fig. III.4. Orbitalii moleculari ai PABM
Ilida a fost preparată prin metoda sării [50,51] și a fost verificată din punctul de vedere al
purității prin mijloace spectrale și chimice. Solvenții, de calitate spectrală, au fost achiziționați de la
Merck și Fluke (Sigma-Aldrich) și utilizați fără purificare suplimentară.
Spectrele electronice de absorbție au fost înregistrate cu spectrofotometrul Specord UV
Vis Carl Zeiss Jena cu sistem de achiziție a datelor.
Parametrii macroscopici ai solvenților (indicele de refracție și permitivitatea electrică),
parametrii Kamlet-Taft ai solvenților și parametrul Reichardt, [66,67] au fost luați de pe
http://www.stenutz.eu/chem/solv26.php. Parametrii solvenților sunt listați în Tabelul III.1.
Tabel III.1. Indicele de refracție, n; permitivitatea electrică, ε; parametrii Kamlet-Taft ai solventului,
π*, α, β; parametrul Reichardt, ; numărul de undă experimental, (cm-1), din maximul benzii
din vizibil a PABM, datorată transferului intramolecular de sarcină
Nr. Solvent n ε π* α β
(cm-1)
1 Dioxan 1.4155 2.20 0.55 0.00 0.37 0.164 22830
2 Tetraclorură de carbon 1.4600 2.24 0.28 0.00 0.10 0.052 22600
3 Benzen 1.5011 2.28 0.59 0.00 0.10 0.111 23400
4 p-Xilen 1.4943 2.28 0.43 0.00 0.12 0.074 23850
5 1,3,5-Trimetilbenzen 1.4980 2.30 0.41 0.00 0.13 0.068 23100
6 Toluen 1.4961 2.34 0.54 0.00 0.11 0.099 23750
7 Anisol 1.5155 4.30 0.73 0.00 0.32 0.198 23400
8 1,2-Dibrometan 1.5389 4.50 0.75 0.00 0.00 0.235 24550
9 Cloroform 1.4460 4.70 0.58 0.20 0.10 0.259 24630
10 n-Butil acetat 1.3950 5.10 0.46 0.00 0.45 0.241 23280
11 Etil acetat 1.4480 6.00 0.55 0.00 0.45 0.228 24330
12 Metil acetat 1.3980 6.70 0.60 0.00 0.42 0.253 24330
13 1,2-Dicloretan 1.3729 10.10 0.81 0.00 0.10 0.327 24500
Page 30
30
14 Octanol 1.4270 10.30 0.40 0.77 0.81 0.537 26350
15 n-Benzil alcool 1.5349 13.30 0.98 0.60 0.52 0.608 26020
16 Ciclohexanol 1.4650 15.00 0.45 0.66 0.84 0.509 26000
17 n-Amil alcool 1.3965 15.20 0.40 0.84 0.86 0.568 26300
18 n-Butil alcool 1.3998 16.00 0.47 0.84 0.84 0.586 25980
19 Metil etil cetonă 1.3793 18.00 0.67 0.06 0.48 0.327 24340
20 Ciclohexanonă 1.4500 18.00 0.76 0.00 0.53 0.281 24770
21 Izobutil alcool 1.3954 18.30 0.40 0.79 0.84 0.552 26300
22 Alcool izopropilic 1.3850 18.30 0.48 0.76 0.84 0.546 25750
23 n-Propil alcool 1.3954 20.40 0.52 0.84 0.90 0.617 26450
24 Acetonă 1.3620 20.70 0.71 0.08 0.43 0.355 24480
25 Alcool etilic 1.3611 26.60 0.54 0.86 0.75 0.654 26760
26 Alcool metilic 1.3288 31.00 0.60 0.98 0.66 0.762 27500
27 Acetonitril 1.3675 36.00 0.75 0.19 0.40 0.460 24600
28 Dimetilformamide 1.4291 37.60 0.88 0.00 0.69 0.386 24480
29 Etilen glicol 1.4300 41.00 0.92 0.90 0.52 0.790 27600
30 Dimetilsulfoxid 1.4740 54.00 1.00 0.00 0.76 0.444 24800
31 Formamidă 1.4555 109.0 0.97 0.71 0.48 0.775 27320
III.3. Rezultate și discuții
III.3.1. Rezultate computaționale
Structura moleculară a PABM a fost obținută utilizând aplicația de modelare moleculară și
chimie computațională Spartan’14 [43,64,65]. Câteva proprietăți atomice și moleculare ale PABM,
calculate cu ajutorul programului Spartan’14, sunt listate în Tabelul III.2.
Tabel III.2. Proprietăți moleculare ale PABM, calculate cu ajutorul programului Spartan
Parametrul
molecular
Valoare QSAR
(Relația cantitativă
structură-activitate)
Valoare
Formula chimică C15H13NO2 Aria 263.02 Å2
Energia -784.026286 ua Volumul 253.24 Å3
Energia (aq) -784.041772 ua Aria suprafeței polare 26.229 Å2
Energia de solvatare -40.66 kJ/mol Ovalitate 1.36
EHOMO -5.24 eV Polarizabilitate 61.18 Å3
Momentul de dipol 3.23 D Numărul donorilor în
legătura de hidrogen
3
Masa moleculară 239.274 uam Numărul acceptorilor în
legătura de hidrogen
0
ELUMO -2.14 eV Temperatura absolută 298.15 K
Fig. III.4 prezintă orbitalii moleculari (orbitali moleculari de valență ocupați și neocupați)
ai PABM. Săgeata din Fig. III.4 indică orientarea momentului de dipol al moleculei.
Page 31
31
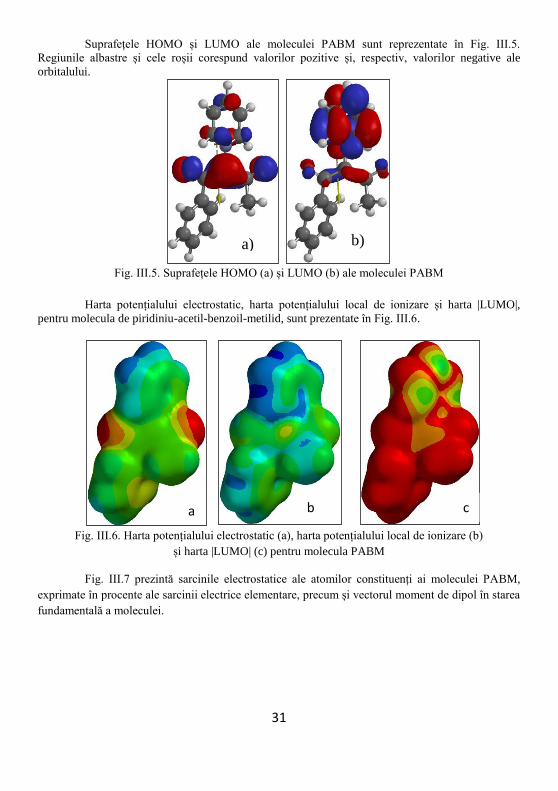
Suprafețele HOMO și LUMO ale moleculei PABM sunt reprezentate în Fig. III.5.
Regiunile albastre și cele roșii corespund valorilor pozitive și, respectiv, valorilor negative ale
orbitalului.
Fig. III.5. Suprafețele HOMO (a) și LUMO (b) ale moleculei PABM
Harta potențialului electrostatic, harta potențialului local de ionizare și harta |LUMO|,
pentru molecula de piridiniu-acetil-benzoil-metilid, sunt prezentate în Fig. III.6.
Fig. III.6. Harta potențialului electrostatic (a), harta potențialului local de ionizare (b)
și harta |LUMO| (c) pentru molecula PABM
Fig. III.7 prezintă sarcinile electrostatice ale atomilor constituenți ai moleculei PABM,
exprimate în procente ale sarcinii electrice elementare, precum și vectorul moment de dipol în starea
fundamentală a moleculei.
a) b)
a b c
Page 32
32
Fig. III.7. Sarcinile electrostatice ale atomilor moleculei PABM
III.3.2. Rezultate spectrale
III.3.2.1. Soluții binare
Numerele de undă măsurate în maximele benzilor de transfer intramolecular de sarcină ale
ilidei studiate, (cm-1), sunt listate în Tabelul III.1. Pe baza Tabelului III.1, datele spectrale au
fost corelate cu parametrii empirici Kamlet-Taft π*, β și α (vezi Fig. III.8) și cu parametrul Reichardt
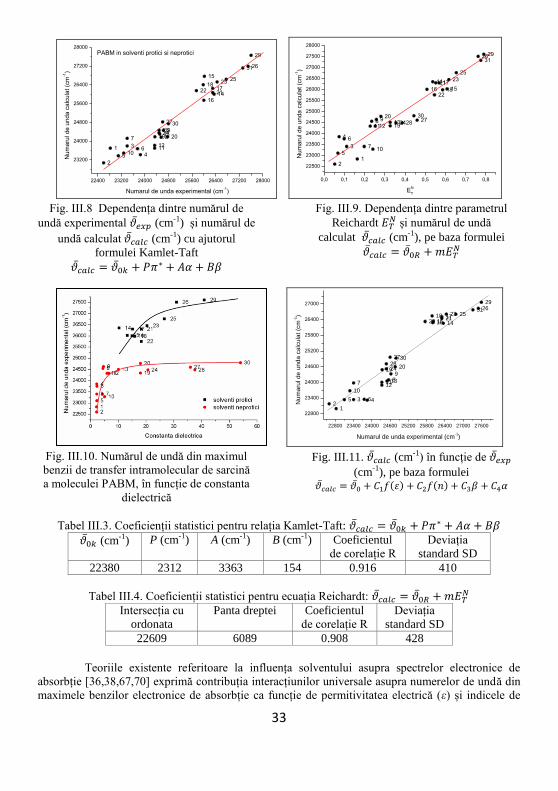
(vezi Fig. III.9). Din Fig. III.8 și tabelele III.3 și III.4, în care parametrii statistici sunt dați pentru
relația Kamlet-Taft, rezultă că PABM este un bun indicator al polarității/polarizabilității solvenților
care interacționează puternic cu solvenții hidroxilici de tipul alcoolilor.
Dependența din Fig. III.9 și coeficienții statistici din tabelele III.3 și III.4 ilustrează o bună
corelație între numerele de undă din maximele benzilor din vizibil ale PABM, datorate transferului
intramolecular de sarcină, și parametrul empiric introdus de Reichardt.
În reprezentarea grafică a numărului de undă experimental (cm-1) în funcție de
constanta dielectrică ε (Fig. III.10), punctele sunt separate pe două curbe: în apropierea curbei
situate la numere de undă mai mici se plasează solvenții neprotici, în timp ce solvenții hidroxilici
sunt situați în apropierea celei de-a doua curbe, la numere de undă mai mari. Distanța dintre cele
două curbe este proporțională cu energia interacțiunilor specifice ale ilidei cu solvenții hidroxilici
[53,56].
Page 33
33
22400 23200 24000 24800 25600 26400 27200 28000
23200
24000
24800
25600
26400
27200
28000
1
2
3
45
6
7 8
9
101112
13
14
15
16
1718
19 20
2122
23
24
25
26
27
28
29
30
31N
um
aru
l d
e u
nd
a c
alc
ula
t (c
m-1)
Numarul de unda experimental (cm-1)
PABM in solventi protici si neprotici
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8
22500
23000
23500
24000
24500
25000
25500
26000
26500
27000
27500
28000
12
3
4
5
6
7
8 9
10
111213
14
151617
18
19
20
21
22
23
24
25
26
2728
29
30
31
Nu
ma
rul d
e u
nd
a c
alc
ula
t (c
m-1)
EN
T Fig. III.8 Dependența dintre numărul de
undă experimental (cm-1) și numărul de
undă calculat (cm-1) cu ajutorul
formulei Kamlet-Taft
Fig. III.9. Dependența dintre parametrul
Reichardt și numărul de undă
calculat (cm-1), pe baza formulei
22800 23400 24000 24600 25200 25800 26400 27000 27600
22800
23400
24000
24600
25200
25800
26400
27000
12
3 45 6
789
10
1112
13
1415161718
1920
2122
23
24
2526
2728
29
30
31
Nu
ma
rul d
e u
nd
a c
alc
ula
t (c
m-1)
Numarul de unda experimental (cm-1)
Fig. III.10. Numărul de undă din maximul
benzii de transfer intramolecular de sarcină
a moleculei PABM, în funcție de constanta
dielectrică
Fig. III.11. (cm-1) în funcție de
(cm-1), pe baza formulei ( ) ( )
Tabel III.3. Coeficienții statistici pentru relația Kamlet-Taft:
(cm-1) P (cm-1) A (cm-1) B (cm-1) Coeficientul
de corelație R
Deviația
standard SD
22380 2312 3363 154 0.916 410
Tabel III.4. Coeficienții statistici pentru ecuația Reichardt:
Intersecția cu
ordonata
Panta dreptei Coeficientul
de corelație R
Deviația
standard SD
22609 6089 0.908 428
Teoriile existente referitoare la influența solventului asupra spectrelor electronice de
absorbție [36,38,67,70] exprimă contribuția interacțiunilor universale asupra numerelor de undă din
maximele benzilor electronice de absorbție ca funcție de permitivitatea electrică (ε) și indicele de
Page 34
34
refracție (n) conform relațiilor (II.2) și (II.3) [42, 71-73]. Deoarece interacțiunile specifice la care
cicloimoniu-ilidele pot participa sunt neglijate în aceste teorii, deplasările totale ale benzilor
electronice sunt descrise de relația (II.1) [74,75].
În relația (II.1), ultimii doi termeni empirici se referă la interacțiunile specifice de tipul
legăturii de hidrogen, în care solventul este partener HBA (acceptor în legătura de hidrogen) sau
HBD (donor în legătura de hidrogen) [64,76]. Coeficienții funcțiilor ce descriu contribuția
interacțiunilor universale depind de parametrii microscopici ai moleculei spectral active, conform
relațiilor (II.12) și (II.13) [49,77].
Ca rezultat al analizei statistice a datelor din Tabelul III.1, următoarea ecuație poate fi
considerată ca descriind valorile numerelor de undă din maximele benzilor de transfer
intramolecular de sarcină ale moleculei PABM:
( ) ( ) ( ) (III.1)
Relația (III.1) a fost obținută cu un coeficient de regresie R = 0.94.
Dependența numărului de undă din maximul benzii de transfer intramolecular de sarcină,
calculat cu relația (III.1), în funcție de valorile experimentale corespunzătoare, este dată de ecuația
(III.2):
( ) (
) (III.2)
Coeficientul de regresie este R = 0.95 și deviația standard, SD = 323 cm-1.
Dependența (cm-1) în funcție de (cm-1) pentru PABM este prezentată în
Fig.III.11. Dependența determinată statistic, de tipul relației (III.1), permite estimarea contribuției
fiecărui tip de interacțiune la deplasarea spectrală înregistrată într-un solvent dat. Calculele au
demonstrat faptul că interacțiunile universale sunt predominante în soluțiile ilidei cu solvenți
neprotici, în timp ce, în aceiași solvenți, interacțiunile specifice sunt nule sau foarte mici. În
solvenții nepolari, interacțiunile de dispersie sunt predominante. În solvenții protici (alcooli),
interacțiunile specifice de tipul legăturilor de hidrogen cresc semnificativ.
Utilizând valorile polarizabilității și momentului de dipol în starea fundamentală (din
Tabelul III.2), valorile coeficienților pentru termenii care descriu contribuția interacțiunilor
universale la deplasările spectrale totale în ecuația (III.1) și dependența lor de parametrii
microscopici ai solutului (vezi relațiile (II.12) și (II.13)), pot fi stabilite următoarele relații:
(III.3)
(III.4)
În relația (III.4), φ este unghiul dintre momentele de dipol ale moleculei PABM în stările
electronice participante la procesul de transfer intramolecular de sarcină.
Discriminantul ecuației (III.4) este mai mare sau egal cu zero pentru unghiuri φ care
satisfac relația φ 80.16 . În aceste condiții, ecuația (III.4) are soluții reale. Când valorile unghiului
din primul cadran sunt modificate din 10 în 10 grade, se obțin datele din Tabelul III.6.
Tabel III.6. Valorile calculate ale momentului de dipol și polarizabilității
în starea excitată a moleculei PABM, pentru diferite unghiuri φ
Nr. φ
(grade)
Ecuația µe (D) αe
(Å3)
1 0 0.0165 µe2 – 6.46 µe + 6.555 = 0 1.02 57.05
2 10 0.0165 µe2 – 6.3619 µe + 6.555 = 0 1.04 56.90
3 20 0.0165 µe2 – 6.070 µe + 6.555 = 0 1.09 56.89
4 30 0.0165 µe2 – 5.5945 µe + 6.555 = 0 1.18 56.86
5 40 0.0165 µe2 – 4.9486 µe + 6.555 = 0 1.32 56.81
6 50 0.0165 µe2 – 4.1524 µe + 6.555 = 0 1.59 56.71
Page 35
35
7 60 0.0165 µe2 – 3.23 µe + 6.555 = 0 2.06 56.47
8 70 0.0165 µe2 – 2.209 µe + 6.555 = 0 3.03 55.80
9 71 0.0165 µe2 – 2.1032 µe + 6.555 = 0 3.20 55.66
10 71,3 0.0165 µe2 – 2.0712 µe + 6.555 = 0 3.257 55.61
11 71,4 0.0165 µe2 – 2.061 µe + 6.555 = 0 3.28 55.59
12 71,5 0.0165 µe2 – 2.0498 µe + 6.555 = 0 3.29 55.58
13 75 0.0165 µe2 – 1.1620 µe + 6.555 = 0 4.10 54.76
14 80 0.0165 µe2 – 1.1217 µe + 6.555 = 0 6.456 51.38
Din Tabelul III.6 se poate observa că momentele de dipol în stare excitată,
corespunzătoare mecanismului de transfer intramolecular de sarcină (care presupune diminuarea
momentelor de dipol în procesul de absorbție a fotonului) sunt obținute pentru 0° φ 71.3°. Acest
fapt reprezintă o limitare a unghiului dintre momentele de dipol în cele două stări electronice
participante la transferul intramolecular de sarcină (TIS) și, de asemenea, pentru momentul de dipol
în stare excitată: 1.02 D µe 3.257 D.
III.3.2.2. Soluții ternare
Pentru a obține informații asupra tăriei interacțiunilor specifice în perechile moleculare de
tipul ilidă – solvent protic, au fost studiate câteva soluții ternare. În solventul binar, cele două lichide
sunt transparente și miscibile, unul protic (1) și unul neprotic (2). Diferența w2 – w1 poate fi estimată
din graficul ln (p1/p2) în funcție de ln(x1/x2), unde x1 este concentrația molară a solventului hidroxilic
(activ din punctul de vedere al interacțiunilor specifice) în întreaga soluție (x1 + x2 = 1) și p1 este
ponderea statistică medie a moleculelor solventului activ în primul strat de solvatare al moleculei
spectral active (p1 + p2 = 1), definită prin [77-83]:
(III.5)
În relația (III.5), este numărul mediu al moleculelor solventului hidroxilic în primul
strat de solvatare și (cm-1) este numărul de undă din maximul benzii de transfer intramolecular de
sarcină (TIS) a ilidei, măsurat în soluții ternare (t), solvent hidroxil (1) și, respectiv, în solvent
neprotic (2).
Rezultatul final [34,40] al modelului celular statistic al soluțiilor ternare este relația (I.29).
Cantitatea w2 – w1 descrie diferența de energie potențială în perechile moleculare de tipul ilidă -
solvent (1) și ilidă - solvent (2). Panta dependenței
în funcție de
este apropiată de 1
atunci când modelul celular statistic este aplicabil soluțiilor ternare studiate. Notăm cu n intersecția
cu ordonata a dreptei (I.29). Valoarea lui n este proporțională cu diferența w2 – w1 care poate fi
estimată [78, 82-84] utilizând relația (III.6):
(III.6)
Să exemplificăm prin soluțiile ternare:
I. PABM + Octanol (1) + 1,2 Dicloretan (2)
Datele experimentale obținute pentru acest set de soluții ternare sunt listate în Tabelul
III.7.
II. PABM + Acid propionic (1) + Cloroform (2)
Datele experimentale obținute pentru acest set de soluții ternare sunt listate în Tabelul
III.8.
Page 36
36
Tabel III.7. Concentrația molară a solventului activ, numărul de undă din maximul
benzii de transfer intramolecular de sarcină și ponderea statistică medie p1 a solventului activ,
pentru soluția ternară PABM + Octanol (1) + 1,2 Dicloretan (2)
Nr.
c1
(%)
x1
(%)
(cm-1)
p1
(%)
1 0 0 - 24500 0.00 -
2 5 2.5 -3.66 24600 0.05 -2.86
3 10 5.2 -2.90 24810 0.17 -1.60
4 25 14.1 -1.81 25230 0.40 -0.43
5 50 33.0 -0.71 25830 0.72 0.94
6 75 59.7 0.39 26220 0.93 2.58
7 100 1.0 - 26350 1.00 -
Tabel III.8. Concentrația molară a solventului activ, numărul de undă din maximul
benzii de transfer intramolecular de sarcină și ponderea statistică medie p1 a solventului activ,
pentru soluția ternară PABM + Acid propionic (1) + Cloroform (2)
Nr. c1
(%)
x1
(%)
(cm-1)
p1
(%)
1 0 0 - 24630 0.00 -
2 5 5.3 -2.88 24700 0.03 -3.45
3 10 10.9 -2.10 24900 0.12 -2.00
4 25 26.3 -1.03 25520 0.39 -0.44
5 50 51.0 0.04 26289 0.73 1.00
6 75 76.3 1.17 26790 0.95 2.98
7 100 100 - 26900 1.00 -
Ponderea statistică medie a solventului activ (1), calculată utilizând formula (III.5), este
reprezentată în Fig. III.12, în funcție de fracția molară a solventului activ, atât pentru soluția (I) cât
și pentru soluția (II). Din Fig. III.12 rezultă o saturare rapidă a primului strat de solvatare al
moleculei PABM cu moleculele solventului activ, pentru ambele seturi de soluții ternare.
Dependența de tipul
în funcție de
este prezentată în Fig. III.13, pentru
ambele seturi de soluții ternare.
Caracteristicile statistice ale liniilor reprezentate în Fig. III.13 sunt date în tabelele III.9 și
III.10. Valorile pantelor celor două linii sunt apropiate de 1 și intersecțiile cu ordonata, bazate pe
relația (III.6), oferă informații despre diferența w2 – w1.
Diferența w2 – w1 a fost estimată din intersecția cu ordonata a dependenței liniare
. Termenii acestei diferențe sunt energiile de interacțiune în perechile
moleculare PABM – moleculele solventului (1) și, respectiv, PABM – moleculele solventului (2).
Din valoarea n a intersecției cu ordonata (Tabelul III.9) rezultă o diferență w2 – w1 =
81.29·10-22 J între energiile de interacțiune în perechile PABM – 1,2 Dicloretan (2) și PABM –
Octanol (1).
Cu datele din Tabelul (III.10) se stabilește valoarea, în jouli, a diferenței w2 – w1 =
47.76·10-22 J în cazul celui de-al doilea set de soluții ternare.
Diferența w2 - w1 este mai mare în perechile moleculare ilidă - octanol, în comparație cu
perechile moleculare ilidă - acid propionic. Acest fapt ar putea indica diferența între mecanismele de
Page 37
37
interacțiune în cele două soluții ternare studiate. Pe de altă parte, octanolul și 1,2 dicloretan sunt
caracterizate prin valori apropiate ale parametrilor macroscopici, în timp ce pentru perechea acid
propionic și cloroform, acești parametri sunt diferiți (vezi Tabelul III.1). În cazul celui de-al doilea
set de soluții ternare, se poate presupune că diferența w2 – w1 conține atât contribuția interacțiunilor
universale, cât și a celor specifice.
0 10 20 30 40 50 60 70 80 90 100 110
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
12
3
4
5
6
7
12
3
4
5
67
PABM+Octanol(1)+1,2-Dicloretan(2)
PABM+Acid propionic(1)+Cloroform(2)
p1
x1 (%)
-4 -3 -2 -1 0 1
-4
-3
-2
-1
0
1
2
3
PABM+Octanol(1)+1,2-Dicloretan(2)
PABM+Acid propionic(1)+Cloroform(2)
ln (
p1/(
1-p
1))
ln (x1/(1-x
1))
Fig. III.12. p1 în funcție de x1 în soluțiile
ternare PABM + Octanol(1) +
1,2Dicloretan(2) și PABM + Acid
propionic(1) + Cloroform(2)
Fig. III.13.
în funcție de
în
soluțiile ternare PABM + Octanol(1) +
1,2Dicloretan(2) și PABM + Acid
propionic(1) + Cloroform(2)
Tabel III.9. Caracteristicile statistice ale liniei
pentru soluția ternară
PABM + Octanol (1) + 1,2 Dicloretan
n ± Δn m ± Δm Date statistice
n Δn m Δm Coeficientul de
corelație R
Deviația standard
SD
1.99 0.11 1.30 0.05 0.99 0.15
Tabel III.10. Caracteristicile statistice ale liniei
pentru soluția ternară
PABM + Acid propionic (1) + Cloroform (2)
n ± Δn m ± Δm Date statistice
n Δn m Δm Coeficientul de
corelație R
Deviația standard
SD
1.10 0.08 1.55 0.05 0.997 0.15
III.4. Concluzii
Analiza cuanto-mecanică a evidențiat natura zwiterionică a ilidei studiate.
În soluțiile PABM au loc atât interacțiuni universale, cât și interacțiuni specifice. În
reprezentarea grafică (cm-1) în funcție de constanta dielectrică, punctele corespunzătoare
solvenților neprotici sunt separate pe o curbă, iar acelea corespunzătoare solvenților protici sunt în
Page 38
38
apropierea unei a doua curbe. La o valoare dată a constantei dielectrice, distanța dintre cele două
curbe este proporțională cu tăria interacțiunilor specifice ale solvenților protici cu ilidele studiate.
Pe baza rezultatelor analizei de regresie liniară multiplă, a fost aproximată contribuția, în
procente, a fiecărui tip de interacțiune. În solvenții neprotici, interacțiunile universale sunt
predominante. În solvenții protici, legăturile de hidrogen dintre gruparea –OH a alcoolilor și
carbanionul moleculei PABM se caracterizează printr-o valoare mare a energie de interacțiune.
Studiul soluțiilor ternare realizate în solvent binar constând într-un solvent protic și unul
neprotic oferă posibilitatea de a estima energia de interacțiune dintre carbanionul ilidei și gruparea –
OH a solventului protic.
Capitolul IV. Momentul de dipol al moleculei de albastru de timol în stare excitată,
estimat prin studii solvatocromice și de mecanică cuantică
IV.1. Introducere
Albastrul de timol este un indicator acido-bazic din familia sulfon-ftaleine, care este
utilizat în mod extensiv pentru determinări volumetrice de punct final. Structura moleculei de
albastru de timol este formată din trei inele benzenice legate de un atom de carbon central, cu o
grupare sulfonică atașată unuia dintre inele și grupări ceto-enolice legate la alte inele (vezi formula
structurală din Fig. IV.1). Denumirea IUPAC a moleculei de albastru de timol este 4-[3-(4-hydroxy-
2-methyl-5-propan-2-ylphenyl)-1,1-dioxobenzo[c]oxathiol-3-yl]-5-methyl-2-propan-2-ylphenol
[86]. Formula chimică este C27H30O5S, are masa molară egală cu 466.592 g/mol, punctul de topire
este cuprins în intervalul 221-224°C iar punctul de aprindere are valoarea de 36°C.
Albastrul de timol este o pulbere cristalină verde-maronie sau cafenie-roșiatică, insolubilă
în apă, dar solubilă în alcooli și soluții alcaline diluate. Molecula are structuri diferite la valori
diferite ale pH-ului.
Fig. IV.1. Formula structurală a
moleculei de albastru de timol
Fig. IV.2. Structura chimică a albastrului de
timol, cu atomii etichetați
Albastrul de timol are multe aplicații în diferite domenii. Astfel, în chimie, albastrul de
timol este utilizat în studiul schimbului de protoni în reacțiile organice [87]. Dezvoltarea unor tipuri
diferite de senzori este aplicația cea mai frecventă a albastrului de timol. În dozimetrie, au fost
preparate dozimetre din peliculă de albastru de timol colorat cu polivinil butiral pentru
monitorizarea radiațiilor ultraviolete sensibile [101]. În medicină, albastrul de timol este utilizat ca
Page 39
39
reactiv colorat în detectarea proteinelor (albumină serică și γ-globulină) [103], sau ca sono-senzor
pentru a studia activitățile sonodinamice și sonocatalitice sub acțiunea iradierii cu ultrasunete, cu
aplicații în terapia sonodinamică și sonocatalitică pentru tratarea tumorilor [104]. Pentru măsurători
gastro-esofagiene, a fost dezvoltat un senzor de pH din fibră optică in vivo, capabil să detecteze pH-
ul în intervalul 1-8, utilizând albastru de timol și albastru de bromfenol ca cromofori [105]. În știința
alimentelor, albastrul de timol este utilizat ca indicator de pH pentru determinarea optică in situ a
pH-ului în timpul tratamentului de înaltă presiune al alimentelor lichide [109].
Scopul acestei lucrări este de a realiza un studiu spectral și cuanto-chimic al albastrului de
timol, pentru a stabili parametrii moleculari ai acestei substanțe în stările electronice responsabile
pentru apariția benzii de absorbție din vizibil. Parametrii electro-optici ai albastrului de timol sunt
indicatori ai reactivității sale și ai abilității de a penetra membranele celulare.
Structura moleculei de albastru de timol este prezentată în Fig. IV.2. Atomii sunt etichetați
pentru a putea exprima ulterior lungimile legăturilor chimice dintre atomii moleculei.
IV.2. Detalii experimentale și computaționale
Molecula de albastru de timol a fost optimizată cu ajutorul programului Spartan’14
[43,66,122]. Au fost utilizate modelele funcționalei de densitate care furnizează rezultate excelente
ale geometriilor de echilibru ale moleculelor organice.
Albastrul de timol și solvenții de calitate spectrală au fost achiziționați de la Sigma-
Aldrich. Spectrele electronice de absorbție ale albastrului de timol au fost înregistrate în 8 solvenți la
temperatura camerei, cu spectrofotometrul QE65000 UV-Vis Ocean Optics. Analiza statistică a
datelor solvatocromice a fost efectuată cu ajutorul programului Origin 8.
IV.3. Rezultate și discuții
IV.3.1. Rezultate computaționale
Proprietățile moleculei de albastru de timol, calculate folosind modelul funcționalei de
densitate EDF2/6-31G* al programului Spartan, sunt listate în Tabelul IV.1.
Tabel IV.1. Proprietățile moleculare ale albastrului de timol Parametri moleculari Valori QSAR
(Relația cantitativă structură-
activitate)
Valori
Formula chimică C27H30O5S Aria (Å2) 477.32
Energia (ua) -1820.17135 Volumul (Å3) 473.58
Energia (aq) (ua) -1820.19335 Aria suprafeței polare (Å2) 82.095
Energia de solvatare (kJ/mol) -57.77 Ovalitatea 1.62
EHOMO (eV) -5.80 Log P 2.54
ELUMO (eV) -2.82 Polarizabilitatea (Å3) 79.09
Momentul de dipol (D) 8.28 Numărul donorilor în legătura
de hidrogen
1
Masa moleculară (uam) 466.598 Numărul acceptorilor în
legătura de hidrogen
2
Tautomeri 5 Temperatura (K) 298.15
Conformeri 192
Page 40
40
Sarcinile electrice ale atomilor moleculei de albastru de timol, calculate cu ajutorul
programului Spartan, sunt afișate în Fig. IV.3, în care săgeata indică orientarea momentului de dipol
al moleculei. Atomii de oxigen ai moleculei de albastru de timol au sarcini electrice negative, fiind
capabili să participe la legături de hidrogen în solvenți hidroxilici.
Programul Spartan poate fi folosit pentru a calcula lungimile legăturilor chimice
interatomice, unghiurile dintre aceste legături și unghiurile diedre. Distanțele cele mai mari sunt
între atomii C5-S1 (1.797 Å), S1-O5 (1.623 Å), C25-C27 (1.538 Å), C15-C17 (1.533 Å) și C15-C16
(1.532 Å), în timp ce între atomii O1-H17 (0.968 Å), O5-H30 (0.983 Å), C4-H3 (1.083 Å), C1-H1,
C3-H2, C6H4, C13-H6 și C23-H19 (1.085 Å) sunt cele mai mici distanțe.
Fig. IV.3. Încărcarea electrostatică a moleculei de albastru de timol
În figurile IV.4 și IV.5 sunt ilustrate câteva reprezentări grafice ale moleculei de albastru
de timol, furnizate de programul Spartan, pe baza unor calcule de chimie cuantică. În Fig. IV.4 sunt
reprezentați orbitalii HOMO și LUMO.
Fig. IV.4. Suprafețele HOMO (a) și LUMO (b) ale moleculei de albastru de timol
a) b)
Page 41
41
Suprafața de densitate a moleculei de albastru de timol, care ilustrează dimensiunea și
forma moleculei, este afișată în Fig. IV.5a. În Fig. IV.5b este reprezentată harta potențialului
electrostatic al moleculei de albastru de timol, hartă care descrie distribuția globală a sarcinii
moleculare, fiind un indicator al adiției electrofile. În Fig. IV.5c este prezentată harta potențialului
de ionizare locală pentru molecula de albastru de timol. Potențialul local de ionizare este un alt
indicator al adiției electrofile. Harta |LUMO| (Fig. IV.5d) este un indicator al adiției nucleofile.
a) b) c) d)
Fig. IV.5. Suprafața de densitate (a), harta potențialului electrostatic (b), harta potențialului de
ionizare locală (c) și harta |LUMO| (d), obținute cu ajutorul metodei funcționalei de densitate
Aria suprafeței polare a moleculelor de albastru de timol este de 82.095 Å2, deci aceste
molecule pot penetra atât membranele celulare, cât și bariera hemato-encefalică.
Valoarea furnizată de programul Spartan pentru coeficientul de partiție, în cazul moleculei
de albastru de timol, demonstrează natura sa hidrofobă (log P = 2.54).
IV.3.2. Rezultate spectrale
Fig. IV.6 prezintă benzile electronice de absorbție ale moleculei de albastru de timol în trei
solvenți: 2-propanol, 1-pentanol și alcool benzilic. Poate fi observată deplasarea benzii moleculare,
în funcție de solventul utilizat.
Fig. IV.6. Benzile electronice de absorbție ale moleculei de albastru de timol
în trei solvenți (2-propanol, 1-pentanol și alcool benzilic)
Numerele de undă experimentale și calculate (utilizând relația II.1)) corespunzătoare
maximelor spectrelor electronice de absorbție, valorile parametrilor macroscopici ai solvenților
utilizați (permitivitatea electrică, ; indicele de refracție, ), funcția de polaritate a solventului ( ), funcția de polarizabilitate a solventului ( ) și valorile parametrilor Kamlet-Taft ai solvenților utilizați ( și ) sunt listate în Tabelul IV.4.
Page 42
42
Tabel IV.4. Numărul de undă experimental, (cm-1) , numărul de undă calculat,
(cm-1), parametrii macroscopici ai solventului (ε, n), funcția de polaritate a solventului f(ε), funcția
de polarizabilitate a solventului f(n) și parametrii Kamlet-Taft ai solventului (α, β) Nr. Solvent
(cm-1)
( ) ( ) (cm-1)
1 Cloroform 18247.18 4.81 0.5595 1.446 0.2667 0.10 0.20 18248.18
2 Alcool
benzilic 18046.63
13.00
0.8000
1.540
0.3138
0.52 0.60
18044.86
3 1-Pentanol 18146.19 13.90 0.8113 1.408 0.2467 0.86 0.84 18146.19
4 2-Butanol 18221.91 16.56 0.8384 1.395 0.2397 0.80 0.69 18230.69
5 1-Butanol 18196.38 17.51 0.8462 1.399 0.2419 0.84 0.84 18188.15
6 2-Propanol 18272.52 19.92 0.8631 1.377 0.2300 0.84 0.76 18254.41
7 1-Propanol 18196.38 21.80 0.8739 1.386 0.2349 0.90 0.84 18213.19
8 Etilenglicol 18247.18 37.70 0.9244 1.429 0.2578 0.52 0.90 18248.67
Coeficientul de corelație al analizei de regresie multiliniară pentru albastrul de timol a fost R =
0.951 iar deviația standard, SD = 15.94 cm-1. Valorile coeficienților de regresie sunt listate în
Tabelul IV.5. Utilizând aceste valori, ecuația (IV.1) poate fi scrisă:
( ) ( ) (IV.1)
După cum se vede în ecuația (IV.1), C1 este pozitiv iar C2 – C4 sunt negativi. Valorile
pozitive ale coeficienților de regresie indică deplasări spectrale spre albastru, în timp ce valorile
negative indică deplasări spectrale spre roșu, odată cu creșterea parametrilor ε, n, β și α ai
solventului.
Tabel IV. 5. Coeficienții regresiei liniare multiple
Intersecția
cu ordonata
C1
(cm-1)
C2
(cm-1)
C3
(cm-1)
C4
(cm-1)
Date statistice
Coeficientul de
corelație R
Deviația
standard SD
18889.42
589.57
-3398.10
-251.34
-198.93
0.951 15.94
Numărul de undă calculat (cm-1) în funcție de numărul de undă experimental
(cm-1) este reprezentat în Fig. IV.7. Dependența liniară dintre numerele de undă calculate și cele
experimentale este caracterizată de panta egală cu 0.979 (eroarea fiind de 0.058). Coeficientul de
corelație este R = 0.976, iar deviația standard SD = 11.15 cm-1. Coeficienții de regresie ai
dependenței liniare dintre valorile experimentale și cele calculate (folosind ecuația (IV.1)) ale
numerelor de undă din maximele benzilor din vizibil ale albastrului de timol sunt listați în Tabelul
IV.6.
Page 43
43
18050 18100 18150 18200 18250 18300
18050
18100
18150
18200
18250 1
2
3
4
5
6
7
8
Nu
ma
rul d
e u
nd
a c
alc
ula
t (c
m-1)
Numarul de unda experimental (cm-1)
Fig. IV.7. (cm-1) în funcție de (cm-1) pentru molecula de albastru de timol
Tabel IV.6. Coeficienții regresiei liniare
Intersecția cu ordonata Panta Date statistice
Valoare Eroare Valoare Eroare Coeficientul de
corelație R
Deviația
standard
379.92 1061.13 0.979 0.058 0.976 11.15
Interacțiunile universale cuantificate prin suma ( ) ( ) predomină în soluțiile albastrului de timol, în timp ce contribuția interacțiunilor specifice la deplasarea spectrală totală nu
depășește 24%, după cum rezultă din calculele efectuate. Dintre interacțiunile universale, ponderea
cea mai mare o au interacțiunile de dispersie.
Relațiile (II.12) - (II.14) ne permit să determinăm momentul de dipol și polarizabilitatea
moleculei de albastru de timol în stare excitată. Pentru albastrul de timol, raza moleculară poate fi
aproximată prin următoarea valoare: a = 2.9765 Å = 2.9765 × 10-8 cm. Cloroformul a fost solventul
utilizat pentru calcularea polarizabilității și a momentului de dipol al moleculei de albastru de timol
în starea excitată. Utilizând ecuațiile (II.12) - (II.15) și valorile C1 = 589.57 cm-1, C2 = -3398.1 cm-1, µg =
8.28 D, αg = 79.09 Å3, Iu = - EHOMO = 5.8 eV (pentru cloroform utilizat ca solvent), s-au obținut
următoarele ecuații:
, (IV.2)
. (IV.3)
Relația (IV.3) are soluții reale atunci când discriminantul este pozitiv:
. (IV.4)
Rezultă că unghiul φ trebuie să satisfacă inegalitatea: φ < 80.28°.
Calculele arată că polarizabilitatea în starea excitată a moleculei de albastru de timol
devine mai mică decât polarizabilitatea în starea fundamentală pentru unghiurile φ cuprinse în
intervalul 27.4° - 27.5°. În acest domeniu unghiular are loc tranziția electronică responsabilă pentru
apariția benzii din domeniul vizibil. Un alt rezultat este acela că, pentru unghiuri mai mari decât 71°,
polarizabilitatea moleculei ar deveni negativă; rezultă că intervalul 71° - 80° trebuie exclus deoarece
soluția nu are sens din punct de vedere fizic.
Fig. IV.8 ilustrează variația momentului de dipol µe și a polarizabilității αe ale moleculei
de albastru de timol în stare excitată, cu unghiul φ (dintre momentele de dipol ale moleculei spectral
active în stările electronice participante la procesul de absorbție).
Page 44
44
0 10 20 30 40 50 60 70 80
-175
-150
-125
-100
-75
-50
-25
0
25
50
75
100
-175
-150
-125
-100
-75
-50
-25
0
25
50
75
100
Po
lari
za
bili
tate
a (
A3)
momentul de dipol
polarizabilitatea
Mo
me
ntu
l d
e d
ipo
l (D
)
Unghiul dintre momentele de dipol in stare fundamentala si stare excitata (grade)
Fig. IV.8. Momentul de dipol și polarizabilitatea moleculei de albastru de timol
în stare excitată, în funcție de unghiul dintre momentele de dipol în stare fundamentală
și în stare excitată
Triunghiul momentelor de dipol asociate procesului de absorbție este desenat în Fig. IV.9.
Variația momentului de dipol poate fi estimată aplicând teorema sinusului. Rezultatele obținute
sunt: φ = 27.50°, µg = 8.28 D, µe = 9.13 D, ξ = 84.84°, ψ = 67.67° și | | = 4.23 D.
Fig. IV.9. Triunghiul momentelor de dipol ale moleculei
de albastru de timol în procesul de absorbție din vizibil
IV.4. Concluzii
În această secțiune au fost calculați, cu ajutorul programului de modelare moleculară
Spartan’14, anumiți parametri electro-optici ai albastrului de timol, pentru a caracteriza reactivitatea
și capacitatea acestuia de a penetra membranele celulare.
Utilizând o metodă variațională în care se calculează momentul de dipol și
polarizabilitatea în stare excitată, pentru diferite valori ale unghiului dintre momentele de dipol ale
moleculei în stările electronice responsabile pentru apariția benzii de absorbție din vizibil, se pot
aproxima (în limitele teoriilor existente referitoare la soluțiile lichide) valorile momentului dipolar
în starea superioară a tranziției electronice. Se observă că, în procesul de absorbție din vizibil,
momentul de dipol al albastrului de timol crește, în timp ce polarizabilitatea moleculei scade.
Variația momentului de dipol al moleculei spectral active, în timpul tranziției electronice, este
aproximativ perpendiculară pe momentul dipolar al moleculei în starea fundamentală.
Page 45
45
Capitolul V. Studiul cuanto-mecanic și spectral al fluoresceinei
V.1. Introducere
Fluoresceina este un colorant de xantină preparat prin încălzirea anhidridei ftalice și
resorcinolului în prezența unui catalizator de zinc, cristalizând sub formă de pulbere de culoare
portocalie sau roșu închis, ușor solubilă în apă și alcool. Adolf von Baeyer a sintetizat-o pentru
prima dată în 1871 [133]. Denumirea IUPAC a fluoresceinei este 3’,6’-dihydroxyspiro[2-
benzofuran-3,9’-xanthene]-1-one, formula chimică este C20H12O5. Are masa molară 332.311 g/mol
și punctul de topire de 315°C.
Structura rigidă mare π-conjugată a fluoresceinei îi conferă câteva proprietăți
spectroscopice speciale, cum ar fi absorbția și emisia intensă a lungimilor de undă din vizibil,
fotostabilitatea ridicată și randamentul cuantic ridicat al fluorescenței [135]. Aceste proprietăți,
împreună cu capacitatea de atașare la anumite specii biomoleculare și ionice, fac ca fluoresceina să
fie un candidat foarte bun pentru o gamă largă de aplicații, dar este cel mai mult utilizată pe scară
largă ca o etichetă fluorescentă datorită costului redus, absorbției ridicate și randamentului cuantic
bun când moleculele de fluoresceină sunt conjugate cu biomolecule [136].
Fluoresceina este utilizată în mod frecvent în biologia celulară, medicină, hidrografie și
criminologie. În medicină, fluoresceina este utilizată în timpul intervențiilor chirurgicale pe cord
deschis, pentru a localiza defecte septale ventriculare musculare. În angiografie, fluoresceina
permite diagnosticarea tulburărilor vasculare. Fluoresceina este, de asemenea, utilizată în chirurgia
tumorilor cerebrale. Unele boli oculare, cum ar fi degenerarea maculară, retinopatia diabetică,
tumorile intravenoase sau aspectele inflamatorii intraoculare pot fi diagnosticate utilizând
fluoresceină. Moleculele atașate fluoresceinei pot ținti unele proteine specifice sau unele structuri
din celule. În medicina legală, fluoresceina este utilizată pentru a detecta urmele de sânge. În
oftalmologie, testul cu fluoresceină este unul auxiliar utilizat pentru evaluarea epiteliului cornean,
pentru a evidenția scurgeri externe ale umorii apoase și pentru a aprecia permeabilitatea barierei
hemato-oculare. Lacrimile curăță ochiul de fluoresceină în câteva minute. În hidrografie,
fluoresceina este utilizată pentru a investiga apele subterane. Substanțele de dispersare sunt
introduse în apă, într-o doză potrivită, iar apa ia culoarea specifică trasorului utilizat. Trasorii pe
bază de fluoresceină au fost utilizați în studiile de urmărire a apelor subterane, în special în
acviferele de apă sărată, unde markerii precum clorura și bromura nu ar putea fi utilizați [156,157].
În chimie, fluorescența este utilizată ca un fotoinițiator eficient în fotopolimerizare [173],
pentru determinarea densității cationice a încărcăturii pe suprafața probei [174] și pentru sintetizarea
dendrimerilor şi dendritelor cationice ale carbosilanului, marcate cu fluoresceină, pentru aplicații
biomedicale [175].
Solvatocromismul descrie modificarea pronunțată a poziției și, uneori, a intensității unei
benzi de absorbție în ultraviolet sau vizibil a solutului, indusă de modificările polarității solventului.
Proprietățile solvatocromice ale fluoresceinei au fost studiate în solvenți apoși și neapoși [135,180-
185], concluzionând faptul că poziția și intensitatea maximelor benzilor de absorbție și fluorescență
sunt în general dependente de polaritatea solventului (în solvenți neprotici) și formarea legăturilor
de hidrogen (în amestecuri binare apoase). Pe baza ecuațiilor Lippert, Bakshiev și Chamma-Viallet,
Acemioğlu et al. au estimat momentele de dipol ale moleculei de fluoresceină în stare excitată [183].
V.2. Detalii experimentale și computaționale
Fluoresceina a fost achiziționată de la Sigma-Aldrich Chemical Company și a fost utilizată
fără purificare suplimentară. Solvenții, de înaltă puritate spectrală, au fost obținuți de la firma
Merck. Formula structurală a fluoresceinei este prezentată în Fig. V.1.
Page 46
46
Fig. V.1 Formula structurală a fluoresceinei
Au fost folosite din literatură numerele de undă pentru fluoresceina dizolvată în cinci
solvenți hidroxilici. În cazul acestora, am presupus că efectul interacțiunilor specifice, reflectat în
deplasările de frecvență, este același pentru toți solvenții, astfel încât, în relația (II.1), ultimii doi
termeni care exprimă contribuția interacțiunilor specifice la deplasarea spectrală au fost eliminați,
urmărind să analizăm doar contribuția interacțiunilor de orientare-inducție și dispersie-polarizare.
Pentru a determina și contribuția interacțiunilor specifice, dar și pentru a îmbunătăți
rezultatele experimentale, am efectuat măsurători cu un număr mai mare de solvenți, adăugând la cei
hidroxilici și solvenți fără gruparea –OH (solvenți neprotici). S-au aplicat calcule de regresie liniară
multiplă datelor din literatură și, ulterior, au fost efectuate măsurători spectrale la o gamă mai largă
de solvenți, atât protici cât și neprotici, în domeniul vizibil și în domenul ultraviolet. Analizăm
rezultatele obținute în următoarele trei tipuri de spectre:
1. spectrele electronice de absorbție ale fluoresceinei, în domeniul vizibil, în cinci
solvenți hidroxilici (methanol, etanol, n-propanol, n-butanol, n-pentanol);
2. spectrele electronice de absorbție ale fluoresceinei, în domeniul ultraviolet, în 14
solvenți (1,4-dioxan, eter dietilic, chloroform, acetat de etil, diclormetan, 1,2-
dicloretan, 1-hexanol, 1-pentanol, 1-butanol, 2-propanol, etanol, methanol, n,n-
dimetilformamidă, n,n-dimetilacetamidă);
3. spectrele electronice de absorbție ale fluoresceinei, în domeniul vizibil, în 16 solvenți
(1,4-dioxan, chloroform, diclormetan, 1-hexanol, 1-pentanol, 2-butanol, 1-butanol, 2-
butanonă, 2-propanol, acetonă, 1-propanol, etanol, n,n-dimetilformamidă, n,n-
dimetilacetamidă, dimetilsulfoxid, formamidă).
Spectrele au fost înregistrate la temperatura camerei, cu spectrofotometrul QE65000 UV-
Vis Ocean-Optics. Programul Origin 8 a fost utilizat pentru a realiza analiza statistică a datelor
solvatocromice. Programul Spartan’14 a fost utilizat pentru modelarea moleculei de fluoresceină și
determinarea unor proprietăți ale acesteia.
V.3. Rezultate și discuții
V.3.1. Rezultate computaționale
Structura chimică a fluoresceinei, optimizată cu ajutorul programul Spartan, este
prezentată în Fig. V.2. Săgeata indică orientarea momentului de dipol al moleculei. Din Fig. V.2
rezultă că, pentru cel mai stabil conformer al moleculei de fluoresceină, ciclul benzofuran este
perpendicular pe ciclul xantină.
Page 47
47
Fig. V.2. Structura chimică a fluoresceinei, optimizată cu ajutorul
programului Spartan'14 (C - gri, O - roșu, H - alb)
Proprietățile moleculare ale fluoresceinei au fost calculate cu ajutorul programului
Spartan'14, utilizând metoda semi-empirică PM3 și, respectiv, modelul funcționalei de densitate
EDF2/6-31G*
În Fig. V.3 sunt afișate sarcinile electrostatice ale atomilor moleculei de fluoresceină
(exprimate în procente ale sarcinii electrice elementare), calculate cu ajutorul programului de
modelare moleculară Spartan, utilizând metoda funcționalei de densitate (Fig. V.3.a) și, respectiv,
metoda semi-empirică PM3 (Fig. V.3.b). Cele mai mari sarcini negative sunt localizate în apropierea
atomilor de oxigen ai moleculei, indicând posibilele interacțiuni specifice ale acestor atomi cu
moleculele alcoolului.
Orbitalii de frontieră ai moleculei de fluoresceină sunt ilustrați în Fig. V.4 (suprafața
HOMO) și, respectiv, în Fig. V.5 (suprafața LUMO).
În figurile V.6 - V.9 sunt afișate reprezentările grafice în spațiu ale densității de electroni,
potențialului electrostatic, potențialului de ionizare locală și |LUMO|.
Structura chimică a fluoresceinei are caracter hidrofil, deoarece log P = - 0.91 < 0. Cu cât
coeficientul de partiție este mai scăzut, cu atât compusul este mai solubil în apă și mai puțin
permeabil prin membrană.
Moleculele de fluoresceină au aria suprafeței polare egală cu 72.3 Å2 dacă se utilizează
metoda funcționalei de densitate, respectiv 75.91 Å2 dacă este utilizată metoda semi-empirică.
Având în vedere aceste valori furnizate de cele două metode, apropiate între ele, rezultă că
moleculele de fluoresceină pot traversa atât membranele celulare, cât și bariera hemato-encefalică.
Acest lucru justifică numărul mare de aplicații ale fluoresceinei în medicină.
Page 48
48
Fig. V.3. Încărcarea electrostatică a moleculei de fluoresceină, calculată prin metoda funcționalei de
densitate a programului Spartan (a) și prin metoda semi-empirică PM3 (b)
Fig. V.4. Suprafața HOMO Fig. V.5. Suprafața LUMO
Fig.V.6.Suprafaț
a densității de
electroni
Fig. V.7. Harta
potențialului
electrostatic
Fig. V.8. Harta
potențialului de
ionizare locală
Fig. V.9. Harta
|LUMO
a) b)
Page 49
49
V.3.2. Rezultate spectrale
Parametrii macroscopici (indicele de refracție și permitivitatea electrică) și parametrii
Kamlet-Taft ai solvenților au fost luați de pe http://www.stenutz.eu/chem/solv26.php.
V.3.2.1. Spectrele electronice de absorbție ale fluoresceinei în solvenți hidroxilici
Numerele de undă din maximele benzilor spectrale, valorile parametrilor macroscopici ai
solvenților (n – indicele de refracție, ε – permitivitatea electrică), funcția de polarizabilitate a
solventului, f(n) și funcția de polaritate a solventului, f(ε) sunt listate în Tabelul V.3.
Tabel V.3. Numerele de undă din maximele spectrelor de absorbție, parametrii macroscopici ai
solventului (n – indicele de refracție, ε – permitivitatea electrică), funcția de polarizabilitate a
solventului f(n) și funcția de polaritate a solventului f(ε)
Solvent (cm-1) ε f(ε) n f(n) (cm-1)
Metanol 21929.82 32.70 0.914 1.328 0.203 21909.38
Etanol 22026.43 24.55 0.887 1.361 0.221 22041.82
n-Propanol 22075.06 20.33 0.866 1.386 0.235 22120.26
n-Butanol 22123.89 17.51 0.846 1.399 0.242 22057.71
n-Pentanol 21739.13 13.90 0.811 1.408 0.247 21752.54
Aceste spectre de absorbție ale fluoresceinei au fost înregistrate în alcooli, în care sunt
posibile interacțiuni specifice prin sarcinile protonilor. Pozițiile benzilor de absorbție în scala
numerelor de undă sunt afectate de posibilele legături de hidrogen dintre grupările –OH ale
alcoolilor și atomii de oxigen ai fluoresceinei. Pentru soluțiile obținute cu alcooli, se poate considera
că interacțiunile specifice afectează în aceeași manieră poziția benzii de absorbție în stabilirea
regresiei multiliniare a deplasărilor spectrale în funcție de funcțiile de polarizabilitate și polaritate
ale solvenților.
A fost utilizată o dependență multiliniară pentru a descrie influența interacțiunilor de
orientare-inducție și dispersie-polarizare asupra deplasărilor spectrale ale fluoresceinei în solvenții
utilizați:
( ) ( ) (V.1)
unde f(ε) și f(n) sunt date de relațiile (II.2) și (II.3).
Datele spectroscopice au fost prelucrate utilizând regresia liniară cu parametri multipli.
Coeficienții relației (V.1) sunt listați în Tabelul V.4.
Tabel V.4. Coeficienții statistici corespunzători relației ( ) ( )
(cm-1)
C1
(cm-1)
C2
(cm-1)
Date statistice
Coeficientul
de corelație R
Deviația
standard SD
5778.22 ± 3511.91 12154.35 ± 2589.92 24780.34 ± 5711.53 0.835 61.783
Astfel, ecuația (V.1) poate fi scrisă:
( ) ( ) (V.2)
Numerele de undă calculate pentru solvenții utilizați sunt prezentate în Tabelul V.3.
Numărul de undă calculat (cm-1) în funcție de numărul de undă experimental (cm-1) este
Page 50
50
reprezentat grafic în Fig. V.10. Există o bună corelație liniară între datele calculate și cele
experimentale ale spectrelor de absorbție ale fluoresceinei, în solvenți hidroxilici (vezi Tabelul V.5).
Tabel V.5. Caracteristicile dependenței liniare dintre
numărul de undă calculat (cm-1) și numărul de undă experimental (cm-1)
Intersecția cu
ordonata (cm-1)
Panta dreptei
Date statistice
Coeficientul de
corelație R
Deviația
standard SD
1597.88 ± 3425.60 0.93 ± 0.16 0.896 47.38
Contribuția fiecărui tip de interacțiune la deplasarea spectrală totală în diferiți solvenți, în
cm-1 și în procente (%), este listată în Tabelul V.6. Semnele pozitive ale parametrilor C1 și C2
determină efectul hipsocromic, adică deplasarea benzilor electronice spre albastru. Deoarece p1 > p2,
rezultă că interacțiunile de orientare-inducție sunt mai puternice decât cele de dispersie-polarizare.
Tabel V.6. Contribuția fiecărui tip de interacțiune universală la deplasarea spectrală totală
Solvent C1f (ε)
(cm-1)
p1
(%)
C2f (n)
(cm-1)
p2
(%)
Metanol 11103.54 68.83 5027.62 31.17
Etanol 10780.98 66.29 5482.62 33.71
n-Propanol 10521.43 64.38 5820.61 35.62
n-Butanol 10285.41 63.18 5994.08 36.82
n-Pentanol 9861.08 61.73 6113.25 38.27
Utilizând expresiile coeficienților de regresie C1 și C2 în funcție de parametrii
microscopici ai moleculei de solut, date de relațiile (II.12) - (II.14), se estimează momentul de dipol
și polarizabilitatea moleculei de fluoresceină în stare excitată. Solventul ales pentru calcul a fost
metanolul. Polarizabilitatea moleculei spectral active în stare excitată poate fi exprimată prin ecuația
(V.3):
(V.3)
Pentru momentul de dipol în stare excitată se obține ecuația:
(V.4)
Ecuația (V.4) are soluții reale atunci când discriminantul este pozitiv:
0 (V.5)
Rezultă că unghiul φ trebuie să satisfacă inegalitatea φ 84.85 . În acest caz, valorile
momentului de dipol în stare excitată sunt:
(V.6)
când unghiul φ variază între 0 φ 84.85 . Din ecuația (V.3) rezultă că αe = 0 când µe = 21.55 D.
Înlocuind µe în ecuația (V.4), se obține φ = 83.54 .
Fig. V.11 ilustrează variația momentului de dipol (µe) și polarizabilității (αe) moleculei de
fluoresceină în stare excitată, cu unghiul φ.
Page 51
51
21700 21750 21800 21850 21900 21950 22000 22050 22100 22150
21700
21750
21800
21850
21900
21950
22000
22050
22100
22150
1
2
3
4
5Nu
ma
rul d
e u
nd
a c
alc
ula
t (c
m-1)
Numarul de unda experimental (cm-1)
banda de absorbtie a fluoresceinei
-10 0 10 20 30 40 50 60 70 80 90 100
-20
-10
0
10
20
30
40
50
60
momentul de dipol
polarizabilitatea
Mom
entu
l de d
ipo
l (D
) / P
ola
rizabili
tate
a (
A3)
Unghiul dintre momentele de dipol in starea fundamentala
si in starea excitata (grade)
Fig. V.10. Numărul de undă calculat în
funcție de numărul de undă experimental
Fig. V.11. Variația momentului de dipol (µe)
și polarizabilității (αe) moleculei spectral
active, cu unghiul φ
V. 3.2.2. Spectrele electronice de absorbție ale fluoresceinei, în ultraviolet și vizibil
Spectrele de absorbție ale fluoresceinei au fost înregistrate în 14 solvenți în domeniul
ultraviolet și în 16 solvenți în domeniul vizibil. Fig. V.12 și Fig. V.13 prezintă câteva dintre benzile
electronice de absorbție ale fluoresceinei.
200 300 400 500 600 7000.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
Abso
rban
ta (
u. a
.)
Lungimea de unda (nm)
1,2-Dicloretan230.03 nm
274.55 nm
300 400 500 600
0.2
0.4
0.6
0.8
1.0
Ab
so
rba
nta
(u
.a.)
Lungimea de unda (nm)
1,4-Dioxan245.81 nm
274.15 nm
200 300 400 500 600 700
0.0
0.4
0.8
1.2
1.6
2.0
Ab
so
rba
nta
(u
.a.)
Lungimea de unda (nm)
1-Butanol234.77 nm
275.72 nm
Fig. VI.12. Spectrele electronice de absorbție ale fluoresceinei, în domeniul ultraviolet
300 400 500 600 7000.4
0.8
1.2
1.6
2.0
Abso
rban
ta (
u.a
.)
Lungimea de unda (nm)
2-Butanol
445.38 nm
487.67 nm
400 500 600
0.2
0.3
0.4
0.5
Abso
rban
ta (
u.a
.)
Lungimea de unda (nm)
2-Butanona
330.17 nm
448.08 nm
300 400 500 600 700
0.5
1.0
1.5
2.0
2.5
3.0
Ab
so
rba
nta
(u
.a.)
Lungimea de unda (nm)
2-Propanol
444.61 nm
485.75 nm
Fig. VI.13. Spectrele electronice de absorbție ale fluoresceinei, în domeniul vizibil
Coeficienții de regresie C1 - C4 din relația (II.1), pentru banda electronică de absorbție a
fluoresceinei în ultraviolet și în vizibil, sunt listați în Tabelul V.10 și, respectiv, în Tabelul V.11.
Page 52
52
Tabel V.10. Coeficienții de regresie obținuți prin analiză de regresie multiliniară
pentru banda electronică de absorbție a fluoresceinei în ultraviolet
Intersecția
cu ordonata
(cm-1)
C1
(cm-1)
C2
(cm-1)
C3
(cm-1)
C4
(cm-1)
Statistică
Coeficientul
de corelație R
Deviația
standard SD
37232.07 ±
164.44
-57.55 ±
78.44
-2797.18
± 651.01
-181.22 ±
55.38
-121.64 ±
39.04
0.826 40.86
Table V.11. Coeficienții de regresie obținuți prin analiză de regresie multiliniară
pentru banda electronică de absorbție a fluoresceinei în vizibil
Intersecția
cu ordonata
(cm-1)
C1
(cm-1)
C2
(cm-1)
C3
(cm-1)
C4
(cm-1)
Statistică
Coeficientul
de corelație R
Deviația
standard SD
25024,78 ±
436,43
201,14 ±
188,95
-12013,51
± 1625,14
-106,59 ±
140,67
224,77 ±
89,87
0,856 103,64
Folosind valorile acestor coeficienți, relația II.11) poate fi scrisă pentru domeniul
ultraviolet (relația (V.7)) și domeniul vizibil (relația (V.8)) după cum urmează:
( ) ( ) ( ) (V.7)
( ) ( ) ( ) (V.8)
Numărul de undă calculat în funcție de numărul de undă experimental este reprezentat
grafic în Fig. V.14 și Fig. V.15. Coeficienții dependenței liniare dintre numărul de undă calculat și
numărul de undă experimental sunt listați în tabelele V.12 și V.13. Datele arată o bună dependență
liniară între valorile experimentale și calculate ale numerelor de undă.
36150 36200 36250 36300 36350 36400 36450 36500
36200
36250
36300
36350
36400
36450
36500
36550
12
3
4 5
6
7
8
9
10
11
12
13
14
Nu
ma
rul d
e u
nd
a c
alc
ula
t (c
m-1)
Numarul de unda experimental (cm-1)
21700 21800 21900 22000 22100 22200 22300 22400 22500 22600 22700
21600
21700
21800
21900
22000
22100
22200
22300
22400
22500
22600
22700
1
2
3
4
5
67
89
10
11
12
13
14
1516
Nu
ma
rul d
e u
nd
a c
alc
ula
t (c
m-1)
Numarul de unda experimental (cm-1)
Fig. V.14. în funcție de (utilizând
relația (V.7))
pentru banda electronică de absorbție a
fluoresceinei, în domeniul ultraviolet
Fig. V.15. în funcție de (utilizând
relația (V.8))
pentru banda electronică de absorbție a
fluoresceinei, în domeniul vizibil
Tabel V.12. Coeficienții dependenței liniare dintre
numărul de undă calculat și cel experimental, în domeniul ultraviolet
Intersecția
cu ordonata
(cm-1)
Panta
Date statistice
Coeficientul de
corelație R
Deviația
standard SD
4377.65 ± 3416.25 0.880 ± 0.094 0.870 33.19
Page 53
53
Tabel V.13. Coeficienții dependenței liniare dintre
numărul de undă calculat și cel experimental, în domeniul vizibil
Intersecția cu ordonata Panta Date statistice
Coeficientul de
corelație R
Deviația standard
SD
2355.79 ± 1824.36 0.894 ± 0.082 0.887 86.82
Interacțiunile de dispersie-polarizare au cea mai mare contribuție la deplasarea spectrală
totală. Ponderea interacțiunilor de dispersie-polarizare este cuprinsă între 66.1% și 92.3% în
domeniul ultraviolet și între 85.5% și 96.9% în domeniul vizibil.
V.3.3. Estimarea momentului de dipol al moleculei de fluoresceină
în stare excitată, utilizând spectrele de absorbție
Utilizând doar datele obținute din spectrele electronice de absorbție, putem apela la o
metodă variațională care ne permite să obținem informații atât despre momentul de dipol electric al
moleculei studiate în stare excitată, cât și despre unghiul dintre momentele de dipol în stările
electronice participante la procesul de absorbție. Raza moleculară a a moleculei de fluoresceină
poate fi aproximată prin valoarea: a = 2.9522 Å = 2.9522·10-8 cm. Pentru efectuarea calculelor de
moment dipolar și polarizabilitate, solventul utilizat a fost cloroformul (Iv = 8.4 eV).
Pentru banda electronică de absorbție a fluoresceinei în domeniul ultraviolet sunt necesare
următoarele date: ; ; ; ;
; (vezi tabelele V.1. și V.10). Se obțin următoarele ecuații:
( )
( ) (V.9)
( ) ( ) (V.10)
Impunând condiția ca discriminantul ecuației (V.10) să fie pozitiv,
, rezultă că valorile reale ale momentului de dipol în stare excitată se obțin atunci când: . Rezultă că unghiul φ dintre momentele de dipol electric
ale moleculei de fluoresceină în cele două stări electronice trebuie să fie mai mic decât 79°.
Considerând valoarea 7.94 D pentru momentul de dipol în starea fundamentală și valoarea
66.33 Å3 pentru polarizabilitatea moleculei de fluoresceină în starea fundamentală, pentru diferite
valori ale unghiului φ se obțin valorile momentului de dipol și polarizabilității în stare excitată.
Pentru , polarizabilitatea moleculei de fluoresceină este mai mică decât polarizabilitatea în starea fundamentală. Acesta este unghiul la care are loc tranziția electronică
corespunzătoare benzii de absorbție din ultraviolet.
Momentele de dipol ale fluoresceinei, corespunzătoare benzii de absorbție din ultraviolet, sunt
reprezentate în Fig. V.16. Se poate utiliza teorema sinusului pentru a rezolva acest triunghi și
obținem:
; și | | . (V.14)
În domeniul vizibil se poate realiza un calcul similar. Folosind datele din tabelele V.1 și
V.11: ; ; ; ;
;
, se obțin următoarele ecuații:
( )
( ) (V.15)
( ) ( ) (V.16)
Page 54
54
Fig. V.16. Triunghiul momentelor
de dipol în stările electronice
participante la tranziția electronică
corespunzătoare procesului de
absorbție
Condiția pentru valorile pozitive ale discriminantului, , dă o valoare a unghiului φ similară cu cea obținută în domeniul ultraviolet: . Pentru , polarizabilitatea moleculei de fluoresceină este mai mică decât polarizabilitatea în starea fundamentală. Acesta este unghiul la care are loc tranziția electronică de
absorbție în domeniul vizibil.
Aplicând teorema sinusurilor în triunghiul momentelor de dipol, ținând cont de relația
și considerând ( ) , rezultă valorile: ; ; | | .
V.4. Concluzii
Molecula de fluoresceină a fost studiată din punct de vedere spectral și cuanto-mecanic
pentru a determina momentul de dipol al moleculei în stare excitată, folosind doar informațiile din
spectrele electronice de absorbție. Atât în ultraviolet cât și în vizibil, tranzițiile au loc cu
creșterea momentului de dipol al moleculei. Momentul de dipol în stare excitată, variația
momentului de dipol în timpul tranziției electronice și unghiul dintre momentul de dipol în stare
fundamentală și în stare excitată sunt mai mari pentru banda electronică de absorbție din domeniul
vizibil, în comparație cu banda din domeniul ultraviolet.
Momentul de dipol în stare excitată pentru o tranziție dată și unghiul dintre momentele de
dipol în stările electronice responsabile pentru procesul de absorbție au fost determinate utilizând o
metodă variațională în care unghiul dintre momentele de dipol a fost variat pas cu pas (din 10 în 10
grade) iar momentul tranziției a fost considerat acela în care polarizabilitatea moleculei de solut
devine mai mică decât polarizabilitatea în starea fundamentală. Prin această metodă, valorile sunt
obținute doar din spectrele electronice de absorbție și sunt în concordanță cu datele din literatură.
Concluzii generale
Cu ajutorul programului de modelare moleculară Spartan, pot fi calculați anumiți
parametri electro-optici ai moleculelor, care caracterizează reactivitatea chimică a acestora
și capacitatea lor de a penetra membranele celulare.
Din studiul cuanto-mecanic rezultă că molecula de quercetină este una hidrofilă, care
poate traversa membranele celulare (având aria suprafeței polare 105,75 Å2), dar nu poate
penetra bariera hemato-encefalică. Aria suprafeței polare este un parametru foarte
important în industria farmaceutică.
Page 55
55
Deplasarea batocromică (spre roșu) a benzii electronice de absorbție din vizibil a
quercetinei, atunci când molecula trece din starea gazoasă în soluții cu polaritate
crescătoare, rezultă din semnele negative ale coeficienților regresiei liniare multiple.
Studiul solvatocromic a subliniat prevalența interacțiunilor de dispersie-polarizare în
soluțiile binare ale quercetinei.
Determinarea parametrilor electro-optici în starea excitată contribuie la o mai bună
reprezentare a transformării suferite de moleculă atunci când interacționează cu radiația
electromagnetică din domeniul vizibil.
În reprezentarea grafică a valorii experimentale a numărului de undă din maximul benzii
electronice de absorbție, în funcție de constanta dielectrică a solventului, punctele
corespunzătoare solvenților neprotici sunt separate pe o curbă, iar acelea corespunzătoare
solvenților protici sunt în apropierea unei a doua curbe. La o valoare dată a constantei
dielectrice, distanța dintre cele două curbe este proporțională cu tăria interacțiunilor
specifice ale solvenților protici cu moleculele spectral active studiate.
În solvenții neprotici predomină interacțiunile universale, în timp ce în solvenții protici,
legăturile de hidrogen se caracterizează printr-o valoare mare a energie de interacțiune.
Studiul soluțiilor ternare realizate în solvent binar constând într-un solvent protic și unul
neprotic oferă posibilitatea de a estima energia de interacțiune dintre molecula spectral
activă (PABM) și gruparea –OH a solventului protic.
Aria suprafeței polare a moleculelor de albastru de timol este de 82.095 Å2, deci aceste
molecule pot penetra atât membranele celulare, cât și bariera hemato-encefalică.
Valoarea furnizată de programul Spartan pentru coeficientul de partiție al moleculei
albastru de timol (log P = 2.54) demonstrează natura sa hidrofobă.
Interacțiunile universale predomină în soluțiile albastrului de timol, în timp ce contribuția
interacțiunilor specifice la deplasarea spectrală totală nu depășește 24%.
Utilizând o metodă variațională în care se calculează momentul de dipol și
polarizabilitatea în stare excitată, pentru diferite valori ale unghiului dintre momentele de
dipol ale moleculei în stările electronice responsabile pentru apariția benzii de absorbție
din vizibil, se pot aproxima valorile momentului dipolar în starea superioară a tranziției
electronice.
Programul Spartan calculează distribuția sarcinii electrice a unei molecule pe atomii
acesteia, distribuție care determină proprietățile fizice și chimice ale moleculei. În cazul
fluoresceinei, cele mai mari sarcini negative sunt localizate în apropierea atomilor de
oxigen, indicând posibilele interacțiuni specifice ale acestor atomi cu moleculele
alcoolului.
Moleculele de fluoresceină (având aria suprafeței polare egală cu 72.3 Å2) pot traversa atât
membranele celulare cât și bariera hemato-encefalică. Acest lucru justifică numărul mare
de aplicații ale fluoresceinei în medicină.
Interacțiunile de dispersie-polarizare din soluțiile fluoresceinei au cea mai mare
contribuție la deplasarea spectrală totală.
Atât în ultraviolet cât și în vizibil, tranzițiile ale fluoresceinei au loc cu creșterea momentului de dipol al moleculei.
Page 56
56
Perspective
Având în vedere rezultatele obținute, îmi propun:
să aplic metoda variațională la mai multe tipuri de molecule spectral active, în
vederea validării ei;
să continui studiul soluțiilor ternare care oferă posibilități mai largi de variație a
parametrilor macroscopici ai solventului (binar) prin alegerea corespunzătoare a
naturii solvenților;
utilizarea modelului statistic al soluțiilor ternare pentru estimarea energiei de
interacțiune dintre două molecule, fapt care nu este posibil prin alte metode.
Bibliografie selectivă
[1] Galeotti, F., E. Barile, P. Curir, M. Dolci, and V. Lanzotti. 2008. Flavonoids from
carnation (Dianthus caryophyllus) and their antifungal activity. Phytochemistry Letters
1:44–48. doi:10.1016/j.phytol. 2007.10.001
[2] Petrus, K., H. Schwartz, and G. Sontag. 2011. Analysis of flavonoids in honey by HPLC
coupled with coulometric electrode array detection and electrospray ionization mass
spectrometry. Analytical and Bioanalytical Chemistry 400 (8):2555–63.
doi:10.1007/s00216-010-4614-7
[3] Smith, C., K. A. Lombard, E. B. Peffley, and W. Liu. 2003. Genetic analysis of quercetin
in onion (Allium cepa L.)’Lady Raider’. Texas Journal of Agriculture and Natural
Resources 16:24–28.
[4] Mitchell, A. E., Y. J. Hong, E. Koh, D. M. Barrett, D. E. Bryant, R. F. Denison, and S.
Kaffka. 2007. Ten-year comparison of the influence of organic and conventional crop
management practices on the content of flavonoids in tomatoes. Journal of Agricultural
and Food Chemistry 55 (15):6154–59. doi:10.1021/jf070344ţ
[5] Kelly, G. S. 2011. Quercetin. Alternative Medicine Review 16 (2):172–94.
[6] D’Andrea, G. 2015. Quercetin: A flavonol with multifaceted therapeutic applications?
Fitoterapia 106:256–71. doi:10.1016/j.fitote.2015.09.018
[7] Perez-Vizcaino, F., D. Bishop-Bailley, F. Lodi, J. Duarte, A. Cogolludo, L. Moreno, L.
Bosca, J. A. Mitchell, and T. D. Warner. 2006. The flavonoid quercetin induces apoptosis
and inhibits JNK activation in intimal vascular smooth muscle cells. Biochemical and
Biophysical Research Communications 346:919–25. doi:10.1016/j.bbrc.2006.05.198
[8] Dower, J. I., J. M. Geleijnse, L. Gijsbers, C. Schalkwijk, D. Kromhout, and P. C. Hollman.
2015. Supplementation of the pure flavonoids epicatechin and quercetin affects some
biomarkers of endothelial dysfunction and inflammation in (pre)hypertensive adults: A
randomized doubleblind, placebo-controlled crossover trial. Journal of Nutrition
145:1459–63. doi:10.3945/jn.115.211888
[9] Bhaskar, S., and A. Helen. 2016. Quercetin modulates toll-like receptor-mediated protein
kinase signaling pathways in oxLDL-challenged human PBMCs and regulates TLR-
activated atherosclerotic inflammation in hypercholesterolemic rats. Molecular and
Cellular Biochemistry 423:53–65. doi:10.1007/s11010-016-2824-9
[10] Jing, Z., Z. Wang, X. Li, X. Li, T. Cao, Y. Bi, J. Zhou, X. Chen, D. Yu, L. Zhu, and S. Li.
2016. Protective effect of quercetin on posttraumatic cardiac injury. Scientific Reports
6:30812. doi:10.1038/srep30812
[11] S.K. Gupta et al. / Materials Science and Engineering C 71 (2017) 919–928
Page 57
57
[12] Psahoulia, F. H., S. Moumtzi, M. L. Roberts, T. Sasazuki, S. Shirasawa, and A. Pintzas.
2007. Quercetin mediates preferential degradation of oncogenic Ras and causes autophagy
in Ha-RAS-transformed human colon cells. Carcinogenesis 28 (5):1021–31.
doi:10.1093/carcin/bgl232
[13] Russo, M., C. Spagnuolo, S. Volpe, A. Mupo, I. Tedesco, and G.-L. Russo. 2010.
Quercetin induced apoptosis in association with death receptors and fludarabine in cells
isolated from chronic lymphocytic leukaemia patients. British Journal of Cancer 103:642–
48. doi:10.1038/sj.bjc.6605794
[14] Srivastava, S., R. R. Somasagara, M. Hegde, M. Nishana, S. K. Tadi, M. Srivastava, B.
Choudhary, and S. C. Raghavan. 2016. Quercetin, a natural flavonoid interacts with DNA,
arrests cell cycle and causes tumor regression by activating mitochondrial pathway of
apoptosis. Scientific Reports 6:24049. doi:10.1038/srep24049
[15] Ferry, D. R., A. Smith, J. Malkhandi, D. W. Fyfe, P. G. deTakats, D. Anderson, J. Baker,
and D. J. Kerr. 1996. Phase I clinical trial of the flavonoid quercetin: Pharmacokinetics
and evidence for in vivo tyrosine kinase inhibition. Clinical Cancer Research 2:659–68.
[16] Fan, P., Y. Zhang, L. Liu, Z. Zhao, Y. Yin, X. Xiao, N. Bauer, J. Gladkich, J. Mattern, C.
Gao, P. Schemmer, W. Gross, and I. Herr. 2016. Continuous exposure of pancreatic
cancer cells to dietary bioactive agent does not induce drug resistance unlike
chemotherapy. Cell Death & Disease 7: e2246. doi:10.1038/cddis.2016.157
[17] Palanivel Rameshthangam et al./ Journal of Materials Science & Technology, Volume 34,
Issue 3, March 2018, Pages 508-522
[18] V.C. George et al./ Journal of Nutritional Biochemistry 45 (2017) 1–14
[19] Rojas, A., J. A. Del Campo, S. Clement, M. Lemasson, M. Garcia-Valdecasas, A. Gil-
Gomez, I. Ranchal, B. Bartosch, J. D. Bautista, A. R. Rosenberg, F. Negro, and M.
Romero-Gomez. 2016. Effect of quercetin on hepatitis C virus life cycle: From viral to
host targets. Scientific Reports 6:31777. doi:10.1038/srep31777
[20] Liu, Z., J. Zhao, W. Li, L. Shen, S. Huang, J. Tang, J. Duan, F. Fang, Y. Huang, H. Chang,
Z. Chen, and R. Zhang. 2016. Computational screen and experimental validation of anti-
influenza effects of quercetin and chlorogenic acid from traditional Chinese medicine.
Scientific Reports 6:19095. doi:10.1038/srep19095
[21] Vedakumari, W. S., N. Ayaz, A. S. Karthick, R. Senthil, and T. P. Sastry. 2017. Quercetin
impregnated chitosan-fibrin composite scaffolds as potential wound dressing materials –
Fabrication, characterization and in vivo analysis. European Journal of Pharmaceutical
Sciences 97:106–12. doi:10.1016/j.ejps.2016.11.012
[22] Cao, L., C. Tan, F. Meng, P. Liu, E. Albert Reece, and Z. Zhao. 2016. Amelioration of
intracellular stress and reduction of neural tube defects in embryos of diabetic mice by
phytochemical quercetin. Scientific Reports 6:21491. doi:10.1038/srep21491
[23] Roslan, J., N. Giribabu, K. Karim, and N. Salleh. 2017. Quercetin ameliorates oxidative
stress, inflammation and apoptosis in the heart of streptozotocin-nicotinamide-induced
adult male diabetic rats. Biomedicine and Pharmacotherapy 86:570–82.
doi:10.1016/j.biopha.2016.12.044
[24] Ren, J., J. Li, X. Liu, Y. Feng, Y. Gui, J. Yang, W. He, and C. Dai. 2016. Quercetin
inhibits fibroblast activation and kidney fibrosis involving the suppression of mammalian
target of rapamycin and β-catenin signaling. Scientific Reports 6:23968.
doi:10.1038/srep23968
Page 58
58
[25] Mehta, V., A. Parashar, A. Sharma, T. R. Singh, and M. Udayabanu. 2017. Quercetin
ameliorates chronic unpredicted stress-mediated memory dysfunction in male Swiss
albino mice by attenuating insulin resistance and elevating hippocampal GLUT4 levels
independent of insulin receptor expression. Hormones and Behavior 89:13–22.
doi:10.1016/j.yhbeh.2016.12.012
[26] Ansari, M. A., H. M. Abdul, G. Joshi, W. O. Opii, and A. Butterfield. 2009. Protective
effect of quercetin in primary neurons against Aβ(1–42): Relevance to Alzheimer’s
disease. Journal of Nutritional Biochemistry 20:269–75.
doi:10.1016/j.jnutbio.2008.03.002
[27] Moreno, L. C. G. e. I., E. Puerta, J. E. Suarez-Santiago, N. E. Santos-Magalhaes, M. J.
Ramirez, and J. M. Irache. 2017. Effect of the oral administration of nanoencapsulated
quercetin on a mouse model of Alzheimer’s disease. International Journal of
Pharmaceutics 517:50–57. doi:10.1016/j. ijpharm.2016.11.061
[28] Singh, T., T. Kaur, and R. K. Goel. 2017. Adjuvant quercetin therapy for combined
treatment of epilepsy and comorbid depression. Neurochemistry International 104:27–33.
doi:10.1016/j. neuint.2016.12.023
[29] McKay, T. B., D. Lyon, A. Sarker-Nag, S. Priyadarsini, J. M. Asara, and D. Karamichos.
2015. Quercetin attenuates lactate production and extracellular matrix secretion in
keratoconus. Scientific Reports 5:9003. doi:10.1038/srep09003
[30] Uthra, C., S. Shrivastava, A. Jaswal, N. Sinha, M. S. Reshi, and S. Shukla. 2017.
Therapeutic potential of quercetin against acrylamide induced toxicity in rats. Biomedicine
& Pharmacotherapy 86:705–14. doi:10.1-016/j.biopha.2016.12.065
[31] Alam, R. T., E. H. Abu Zeid, and T. S. Imam. 2017. Protective role of quercetin against
hematotoxic and immunotoxic effects of furan in rats. Environmental Science and
Pollution Research 24:3780–89. doi:10.1007/s11356-016-8108-9
[32] Environ Sci Pollut Res, DOI 10.1007/s11356-016-8108-9
[33] Chondrogianni, N., S. Kapeta, I. Chinou, K. Vassilatou, I. Papassideri, and E. S. Gonos.
2010. Antiageing and rejuvenating effects of quercetin. Experimental Gerontology
45:763–71. doi:10.1016/j. exger.2010.07.001
[34] Environ Sci Pollut Res, DOI 10.1007/s11356-016-8108-9
[35] A.A. Lozano-Pérez et al. / International Journal of Pharmaceutics 518 (2017) 11-19
[36] McRae, E. G. 1957. Theory of solvent effects on molecular electronic spectra. Frequency
shifts. Journal of Physical Chemistry 61 (5):562–572.
[37] Abe, T. 1965. Theory of solvent effects on molecular electronic spectra. Frequency shifts.
Bulletin of the Chemical Society of Japan 38 (8):1314–18. doi:10.1246/bcsj.38.1314
[38] Bakhshiev, N. G. 1972. Spectroscopy of intermolecular interactions. Leningrad: Nauka (in
Russian).
[39] Kamlet, M. J., J. L. M. Abboud, M. H. Abraham and R. W. Taft. 1983. Linear solvation
energy relationships. 23. A comprehensive collection of the solvatochromic parameters,
π*, α, and β, and some methods for simplifying the generalized solvatochromic equation.
Journal of Organic Chemistry 48 (17):2877–87. doi:10.1021/jo00165a018
[40] Rusu, E., D. O. Dorohoi, and A. Airinei. 2008. Solvatochromic effects in the absorption
spectra of some azobenzene compounds. Journal of Molecular Structure 887 (1–3):216–
19. doi:10.1016/j. molstruc.2008.01.053
[41] Dimitriu, M., D. G. Dimitriu, and D. O. Dorohoi. 2008. Supply to the spectral shifts of
each type of intermolecular interactions in binary solutions. Optoelectronics and Advanced
Materials 2 (12):867–70.
Page 59
59
[42] Dorohoi, D. O., D. G. Dimitriu, M. Dimitriu, and V. Closca. 2013. Specific interactions in
N-ylid solutions, studied by nuclear magnetic resonance and electronic absorption
spectroscopy. Journal of Molecular Structure 1044:79–86.
doi:10.1016/j.molstruc.2012.12.039
[43] Hehre, W. J. 2003. A guide to molecular mechanics and quantum chemical calculations.
Irvine, CA, USA: Wavefunction Inc.
[44] Fleming, I. 1976. Frontier orbitals and organic chemical reactions. London: John Wiley &
Sons.
[45] Comer, J., and K. Tam. 2001. Lipophilicity profiles: Theory and measurement. In
Pharmacokinetic optimization in drug research: Biological, physicochemical, and
computational strategies, ed. B. Testa, H. van de Waterbeemd, G. Folkers, and R. Guy,
275–304. Weinheim: Wiley-VCH.
[46] Parthasarathi, R., V. Subramanian, D. R. Roy, and P. K. Chattaraj. 2004. Electrophilicity
index as a possible descriptor of biological activity. Bioorganic & Medicinal Chemistry 12
(21):5533–43. doi:10.1016/j.bmc.2004.08.013
[47] Hitchcock, S. A., and L. D. Pennington. 2006. Structure – Brain exposure relationships.
Journal of Medicinal Chemistry 49 (26):7559–83. doi:10.1021/jm060642i
[48] Benchea, A. C., D. Babusca, D. G. Dimitriu, and D. O. Dorohoi. 2017. Quantum-
mechanical study and spectral analysis on some derivatives of Rhodamine in solutions.
Spectrochimica Acta Part A 172:91–99. doi:10.1016/j.saa.2016.04.027
[49] Babusca, D., A. C. Benchea, D. G. Dimitriu, and D. O. Dorohoi. 2016. Solvatochromic
characterization of Sudan derivatives in binary and ternary solution. Analytical Letters 49
(16):2615–26. doi:10.1080/00032719.2016.1152275
[50] I. Zugravescu, M. Petrovanu, N-Ylid Chemistry, McGraw Hill, Acad. Press, N.Y. London,
1976.
[51] Johnson, N-Ylid Chemistry, Academic Press, New York, 1966.
[52] F. Kröhnke, Synthesis using pyridinium salts. 6. Specific synthesis of pyridines and
oligopyridines, Synthesis-Stuttgart 1 (1976) 1-24.
[53] D.O. Dorohoi, Electronic spectroscopy of N-Ylids, J. Mol. Struct. 704 (1–3) (2004) 31-43.
[54] D.O. Dorohoi, D.H. Partenie, L.M. Chiran, C. Anton, About the electronic absorption
spectra (EAS) and electronic diffusive spectra (EDS) of some pyridazinium ylids, J. Chim.
Phys. Phys.-Chim. Biol. 91 (3) (1994) 419–431.
[55] D.O. Dorohoi, G. Surpateanu, D. Iancu, Intermolecular interactions in some pyridinium
ylids solutions, An. Stiint. Univ. Al.I.Cuza Iasi, s. Ib Fizica 30 (1983) 59–64.
[56] C. Gheorghies, L.V. Gheorghies, D.O. Dorohoi, Solvent influence on some complexes
realized by hydrogen bond, J. Mol. Struct. 887 (1–3) (2008) 122–127.
[57] A. Pawda, 1, 3 Cycloaddition Reactions, Wiley Interscience, NY, 1984.
[58] V. Melnig, I. Humelnicu, D.O. Dorohoi, Thermal dimerization kinetics of 3-(pbromo)
pyridazinium benzoyl methylid in solutions, In. J. Chem. Kinet. 40 (50) (2008) 230–239.
[59] M. Petrovanu, I. Zugravescu, Cycloaddition Reactions, Academic Press, Bucuresti, 1987.
[60] L. Leontie, I. Olariu, I.G. Rusu, On the charge transport in some new carbanion
disubstituted ylids in thin films, Mater. Chem. Phys. 80 (2) (2003) (5-6-511).
[61] L. Leontie, M. Roman, F. Branza, C. Podaru, I.G. Rusu, Electrical and optical properties
of some new synthesized ylids in thin films, Synth.Mat. 138 (1–2) (2003) 157–163.
[62] M. Ungureanu, I.Mangalagiu,M. Petrovanu, Pyridaziniumylures in Pharmacy I, Ann.
Pharm. Fr. 55 (2) (1997) 69–77.
[63] I. Mangalagiu, G. Mangalagiu, M. Roman, M. Caprosu, M. Petrovanu, Pyridazinium
ylures in Pharmacy II, Ann. Pharm. Fr. 58 (1) (2000) 86–92.
Page 60
60
[64] David Young, Computational Chemistry, Appendix A. A.1.6, Wiley-Interscience, 2001
330 (SPARTAN).
[66] Spartan'14 for Windows, Macintosh and Linux, Tutorial and User's Guide, January 10,
2014 Modeling, 2014.
[66] a) M.J. Kamlet, J.L.M. Abboud, M.H. Abraham, R.W. Taft, Linear solvation energy
relation shifts. 23. A comprehensive collection of the solvatochromic parameters, pi star,
alpha and beta and some methods for simplifying the generalized solvation equation, J.
Organomet. Chem. 48 (1983) 2877–2887; b) R.W. Taft, J.L.M. Abboud, M.J. Kamlet,
M.H. Abraham, Linear solvation energy relations, J. Solut. Chem. 14 (3) (1985) 153–186.
[67] C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, VCH Publications,
Weiheim, 1988.
[68] T. Schlick, Molecular Modeling and Simulation; An Interdisciplinary Guide, Springer
Verlag, New York, 2002.
[69] H.D. Holtjie, W. Sippl, D. Rognan, C. Folkers, Molecular Modeling, Wiley, VCH, New
York, 2003.
[70] V. Closca, L.M. Ivan, D.O. Dorohoi, Intermolecular interactions in binary and ternary
solutions of two cycloimmonium carbethoxy-anilido-methylids, Spectrochim. Acta A
Mol. Biomol. Spectrosc. 122 (2014) 670–675.
[71] V. Pop, D.O. Dorohoi, V. Holban, Molecular interactions in binary solutions of 4-
aminophthalimide and 3-p-cumyl-pyridazinium-acetyl-benzoyl methylid, Spectrochim.
Acta A 50 (14) (1994) 2281–2289.
[72] M. Homocianu, A. Airinei, D.O. Dorohoi, Intermolecular interactions in binary solutions
of some pyridazinium ylids studied by visible electronic spectroscopy, Proceedings of
SPIE, vol.8001, 2013 Article 80013K, Published 2011, http://dx.doi.
org/101117/12.891948.
[73] C. Nadejde, D.E. Creanga, I. Humelnicu, D.O. Dorohoi, Study of intermolecular
interactions in rifampicin ternary solutions. Calculation of microscopic parameters of
rifampicin molecules, J. Mol. Liq. 150 (1–3) (2009) 51–55.
[74] E.R.Stanculescu L. Stroia D.O. Dorohoi, Solvatochromism used in determining some
electro-optical parameters, Proceedings SPIE, vol.8001, 2017 nr. Art. 80012S Published in
2011.
[75] D. Babusca, A.C. Morosanu, D.O. Dorohoi, Spectral study of interactions between
zwitterionic compounds and protic solvents, Spectrochim. Acta A Mol. Biomol.
Spectrosc. 172 (SI) (2017) 58–64.
[76] A.C. Benchea, D. Babusca, D.G. Dimitriu, D.O. Dorohoi, Spectral study of Rhodamine
dyes in binary solutions, J. Mol. Struct. 1140 (SI) (2017) 71–76.
[77] Y.T. Mazurenko, Universalnie Vzaimodeistvii trex componentov jidkostei, (Universal
interactions in ternary solutions) Opt. I Spektrosk. T. XXXIII, vol. 7, 1972 1060–1064.
[78] V. Pop, D.O. Dorohoi, M. Delibas, Considerations on the statistic model of the
intermolecular interactions in ternary solutions, An. Stiin. Univ. Al. I. Cuza, Iasi, t. XXXII
s. Ib, Fizica (1986) 79–84.
[79] D. Dorohoi, V. Pop, The electronic spectra shifts of some cycloimmonium ylids in ternary
solutions, An. Stiin. Univ. Al. I. Cuza, Iasi, t. XXXIII s. Ib, Fizica (1987) 78–86.
[80] D.O. Dorohoi, M. Avadanei, M. Postolache, Characterization of the solvation spheres of
some dipolar spectrally active molecules in binary solvents, Optoelectron. Adv. Mater.
Rapid Commun. 2 (8) (2008) 51–55.
Page 61
61
[81] M. Avadanei, M.L. Ivan, C. Nadejde, D.O. Dorohoi, Spectral and thermodynamical
studies on iso-Qinolinium-carbethoxy-methylid (iQCEM) solutions with binary solvent
water (W) and ethanol (E), Rev. Chim. (Bucharest, Rom.) 66 (2) (2015) 201–204.
[82] M.M. Dulcescu, C. Stan, D.O. Dorohoi, Spectral investigations on the influence of the
hydroxyl solvents on intermolecular interactions in some pyridinium ylids ternary
solutions, Rev. Chim. (Bucharest, Rom.) 61 (2) (2010) 1219–1222.
[83] M.M. Dulcescu, C. Stan, D.O. Dorohoi, Study of intermolecular interactions in
waterethanol solutions of some pyridazinium ylids, Rev. Roum. Chim. 55 (7) (2010) 403–
408.
[84] M. Avadanei, D.O. Dorohoi, Interaction energy in pairs of pyridazinium ylid – solvent
molecules estimated by spectral means within the cell ternary solution model, Ukr. J.
Phys. 57 (2) (2012) 118–122.
[85] P. Balderas-Hernández, R. Vargas, A. Rojas- Hernández, M. T. Ramírez-Silva, M.
Galván, Dimerization of thymol blue in solution: Theoretical evidence, Talanta 71 (2007)
1061-1067.
[86] PubChem Database, https://pubchem.ncbi.nlm.nih.gov, accesed at 17 April 2018.
[87] R. Maggini, F. Secco, M. Venturini, Dynamic analysis of chemical relaxation effects: A
study of salicyclate-indicator systems by a T-jump apparatus with laser monitoring, J.
Phys. Chem. A 101 (1997) 5666-5671.
[88] T. Fujii, K. Toriumi, Ultraviolete-visible absorption spectra of Thymol blue curing the sol-
gel-xerogel transition of Tetraethyl orthosilicate: In situ probe of microchemical
environment, J. Chem. Soc. Faraday Trans. 89(18) (1993) 3437-3441.
[89] R. Müller, N. Anders, J. Titus, D. Enke, Ultra-thin porous glass membranes – An
innovative material for the immobilization of active species for optical chemosensors,
Talanta 107 (2013) 255-262.
[90] S. Capel-Cuevas, M.P. Cuéllar, I. de Orbe-Payá, M.C. Pegalajar, L.F. Capitán-Vallvey,
Full range optical pH sensor based on imaging techniques, Anal. Chim. Acta 681 (2010)
71-81.
[91] S. Capel-Cuevas, M.P. Cuéllar, I. de Orbe-Payá, M.C. Pegalajar, L.F. Capitán-Vallvey,
Full range optical pH sensor array based on neural networks, Microchem. J. 97 (2011)
225-233.
[92] A. Martinez-Olmos, S. Capel-Cuevas, N. López-Ruiz, A.J. Palma, I. de Orbe, L.F.
Capitán-Vallvey, Sensor array-based optical portable instrument for determination of pH,
Sens. Actuators B-Chem. 156 (2011) 840-848.
[93] V.M.C. Rérolle, C.F.A. Floquet, A.J.K. Harris, M.C. Mowlem, R.R.G.J. Bellerby, E.P.
Achterberg, Development of a colorimetric microfluidic pH sensor for autonomous
seawater measurements, Anal. Chim. Acta 786 (2013) 124-131.
[94] H. Zhang, R.H. Byrne, Spectrophotometric pH measurements of surface seawater at in-
situ conditions: absorbance and protonation behavior of thymol blue, Mar. Chem. 52
(1996) 17-25.
[95] L.M. Mosley, S.L.G. Husheer, K.A. Hunter, Spectrophotometric pH measurement in
estuaries using thymol blue and m-cresol purple, Mar. Chem. 91 (2004) 175-186.
[96] V.M.C. Rérolle, C.F.A. Floquet, M.C. Mowlem, R.R.G.J. Bellerby, D.P. Connelly, E.P.
Achterberg, Seawater-pH measurements for ocean-acidification observations, Trends
Anal. Chem. 40 (2012) 146-157.
[97] B. Yang, M.C. Patsavas, R.H. Byrne, J. Ma, Seawater pH measurements in the field: A
DIY photometer with 0.01 unit pH accuracy, Mar. Chem. 160 (2014) 75-81.
Page 62
62
Anexe
Lista lucrărilor publicate în reviste cotate ISI
1. D. Babusca, C. Morosanu, D. O. Dorohoi, Spectral Study of Specific Interactions
Between Zwitterionic Compounds and Protic Solvents, Spectrochimica Acta A, 172, 58-
64, 2017
2. A. C. Benchea, D. Babusca, A. C. Morosanu, D. G. Dimitriu, D. O. Dorohoi, Spectral
study of Rhodamine dyes in binary solutions, Journal of Molecular Structure,1140, 71-76,
2017
3. C. Morosanu, A.C. Benchea, D. Babusca, D.G. Dimitriu, D.O. Dorohoi, Quantum
Mechanical Characterization And Solvatochromic Study Of Quercetine, Analytical
Letters, http://dx.doi.org/10.1080/00032719.2017.1291657
4. A. C. Morosanu, A. Gritco, D. E. Creanga, D. O. Dorohoi, Computational and
Solvatochromic study of pyridinium-acetyl-benzoyl-methylid (PABM), Spectrochimica
Acta A, 189, 307–315, 2018
5. D. Babusca, A. C. Morosanu, A. C. Benchea, D. G. Dimitriu, D. O. Dorohoi, Spectral
and quantum mechanical study of some azo-derivatives, Journal of Molecular Liquids,
https://doi.org/10.1016/j.molliq.2018.03.125
Lucrări trimise spre publicare în reviste cotate ISI
1. C. Morosanu, D. Babusca, A. C. Benchea, D. G. Dimitriu, D. O. Dorohoi, Excited state
dipole moment of thymol blue, estimated from solvatochromic and quantum-mechanical
studies, trimis la Spectrochimica Acta A
2. A. C. Morosanu, D. G. Dimitriu, D. O. Dorohoi, Excited state dipole moment of
fluorescein molecule estimated from electronic absorption spectra, trimis la Journal of
Molecular Structure
Lucrări publicate în reviste proceedings ISI
1. D. Babusca, A. C. Benchea, A. C. Morosanu, D. G. Dimitriu, and D. O. Dorohoi, Solvent
empirical scales and their importance for the study of intermolecular interactions, AIP
Conference Proceedings, 1796, 030011, 2017, doi: 10.1063/1.4972376.
2. A. C. Benchea, D. Babusca, A. C. Morosanu, D. G. Dimitriu, D. O. Dorohoi, Excited
state molecular polarizability estimated by solvatochromic means, AIP Conference
Proceedings, 1796, 030013 ,2017, doi: 10.1063/1.4972378.
3. A. C. Morosanu, A. Gritco, D. G. Dimitriu, D. O. Dorohoi, C. Cheptea Quantum
Mechanical and Spectral Study of Fluorescein, Publicat în: 2017 E-Health and
Bioengineering Conference (EHB), July 2017, doi: 10.1109/EHB.2017.7995500
Lucrări publicate in Reviste BDI/B+
1. D. Babusca, C. A. Morosanu, D. O. Dorohoi, Solvatochromic study of two pyridazinium
ilids binary solutions, International Conference Knowledge-Based Organization Vol.
XXII No 3 2016.
Page 63
63
2. C. Morosanu, D. O. Dorohoi, Spectral Means to Estimate the Energy of Internal
Interactions in Liquid Solutions, Buletinul Institutului Politehnic din Iaşi,
http://www.agir.ro/buletine/2407.pdf
3. C. Morosanu, V. Closca, N. Melniciuc Puica, D. O. Dorohoi, Interaction Energy in Pairs
of 4’-Aryl-1, 2, 4-Triazol-1-Ium-(P)-Chloro-Phenacylids and Hydroxyl Solvents,
Buletinul Institutului Politehnic din Iaşi, http://www.agir.ro/buletine/2416.pdf
4. A. Gritco-Todirașcu, A. C. Moroșanu, L. M. Ivan, C. Cheptea, D. Dorohoi,
Computational Study of Two Carbanion Mono-Substituted Pyridazinium Ylids, Buletinul
Institutului Politehnic din Iaşi, volumul 62 (66), numărul 4, 2016.
5. Quantum Mechanical Characterization of Thymol Blue, A. C. Moroșanu, D. G. Dimitriu,
D. O. Dorohoi, Buletinul Institutului Politehnic din Iaşi, volumul 64 (68), numărul 2, 2018
Articole publicate în reviste de categoria C și D
1. C. Moroșanu, Metode spectrale pentru studierea interacțiunilor intermoleculare în soluții,
volumul Concursului Naţional de Referate şi Comunicări Ştiinţifice – cu participare
internațională - „Ştefan Procopiu”, Piatra-Neamţ, mai 2015.
2. A. C. Moroșanu (Călugăru), A. Grițco (Todirașcu), D. O. Dorohoi, Investigarea
substanțelor cu ajutorul metodelor spectrale și modelării computaționale, volumul
Concursului Naţional de Referate şi Comunicări Ştiinţifice – cu participare internațională -
„Ştefan Procopiu”, Piatra-Neamţ, aprilie 2016
3. C. Călugăru (Moroșanu), D. O. Dorohoi, Spectre electronice de absorbție ale unor
compuși organici cu efecte farmaceutice, volumul Concursului Naţional de Referate şi
Comunicări Ştiinţifice – cu participare internațională - „Ştefan Procopiu”, Piatra-Neamţ,
mai 2017
4. A. C. Morosanu, M. Avadanei, V. Closca, D. O. Dorohoi, Statistical cell model of
ternary solutions applied for determining the interaction energy between two molecules,
volumul Concursului Naţional de Referate şi Comunicări Ştiinţifice – cu participare
internațională - „Ştefan Procopiu”, Piatra-Neamţ, mai 2018
Participări Conferințe Internaționale
a) Lucrări prezentate oral
1. Spectroscopie moleculară aplicată în biologie, chimie şi medicină, A. C. Călugăru-
Moroşanu, Colocviul Internațional de Fizică Evrika Cygnus, 28-30 august 2015, Iași,
România.
2. Quantum Mechanical and Spectral Study of Fluorescein, A. C. Morosanu, A. Gritco, D.
G. Dimitriu, D. O. Dorohoi, C. Cheptea, The 6th IEEE International Conference on E-
Health and Bioengineering - EHB 2017, Grigore T. Popa University of Medicine and
Pharmacy, June 22-24, 2017, Sinaia, Romania
b) Lucrări prezentate poster
1. Spectral Means to Estimate the Energy of Internal Interactions in Liquid Solutions, C.
Morosanu, D. O. Dorohoi, IBWAP 2015, The 15th International Balkan Workshop on
Applied Physics, July 2-4, 2015, Constanta, Romania
Page 64
64
2. Spectral and Quantum Mechanical Studies of Dimerization Reaction of Some Carbanion
Monosubstituted Pyridazinium Ylids, C. Morosanu (Calugaru), D. Babusca, D. O.
Dorohoi, Colloquium Spectroscopicum Internationale XXXIX, 30.08 – 03.09. 2015,
Figueira da Foz, Portugalia
3. Internal energy in liquids estimated by spectral means, A C Călugăru (Moroșanu), D. O.
Dorohoi, 10th International Conference Processes in Isotopes and Molecules, 23-25
september 2015, Cluj-Napoca, Romania
4. Specific and universal interactions in solutions of some zwitterionic compounds, A. C.
Calugaru (Morosanu), A. Gritco (Todirascu), D. E. Creanga, D. O. Dorohoi, 33rd
European Congress on Molecular Spectroscopy, 30 July - 4 August 2016, Szeged,
Hungary
5. Quantum Mechanical Characterization And Solvatochromic Study Of Quercetine, C.
Morosanu, A.C. Benchea, D. Babusca, D.G. Dimitriu, D.O. Dorohoi, International
Conference on Analytical and Nanoanalytical Methods for Biomedical and Environmental
Sciences, June 29th-July 1st, 2016, Braşov
Participări Conferințe Naționale
a) Lucrări prezentate oral
1. Interaction Energy in Pairs of 4’-Aryl-1, 2, 4-Triazol-1-Ium-(P)-Chloro-Phenacylids and
Hydroxyl Solvents, C. Morosanu, V. Closca, N. Melniciuc Puica, D. O. Dorohoi, The
20th National Conference on Thermodynamics with International Participation, June 4-5,
2015, Iasi, Romania
2. Spectral Means to Estimate the Energy of Internal Interactions in Liquid Solutions, C.
Morosanu, D. O. Dorohoi, The 20th National Conference on Thermodynamics with
International Participation, June 4-5, 2015, Iasi, Romania
3. Metode spectrale pentru studierea interacțiunilor intermoleculare în soluții, C. Moroșanu,
Concursul Naţional de Referate şi Comunicări Ştiinţifice – cu participare internațională -
„Ştefan Procopiu”, mai 2015, Piatra-Neamţ.
4. Investigarea substanțelor cu ajutorul metodelor spectrale și modelării computaționale, A.
C. Moroșanu (Călugăru), A. Grițco (Todirașcu), D. O. Dorohoi, Concursul Naţional de
Referate şi Comunicări Ştiinţifice – cu participare internațională - „Ştefan Procopiu”,
aprilie 2016, Piatra-Neamţ.
5. Spectre electronice de absorbție ale unor compuși organici cu efecte farmaceutice, C.
Călugăru (Moroșanu), D. O. Dorohoi, Concursul Naţional de Referate şi Comunicări
Ştiinţifice – cu participare internațională - „Ştefan Procopiu”, mai 2017, Piatra-Neamţ,
6. Statistical cell model of ternary solutions applied for determining the interaction energy
between two molecules, A. C. Morosanu, M. Avadanei, V. Closca, D. O. Dorohoi,
Concursul Naţional de Referate şi Comunicări Ştiinţifice – cu participare internațională -
„Ştefan Procopiu”, Piatra-Neamţ, mai 2018
b) Lucrări prezentate poster
1. Method for estimating the domain of variation of the excited electric dipole moment from
electronic absorption spectra, C. Morosanu, D. Babusca, A. C. Benchea, D. G. Dimitriu,
D. O. Dorohoi, Conferința Națională de Fizică Aplicată, 26-27 noiembrie 2016, Iași